

Using hydrogen deuterium exchange mass spectrometry to engineer optimized constructs for crystallization of protein complexes: Case study of PI4KIIIβ with Rab11
- PMID: 26756197
- PMCID: PMC4832280
- DOI: 10.1002/pro.2879
Using hydrogen deuterium exchange mass spectrometry to engineer optimized constructs for crystallization of protein complexes: Case study of PI4KIIIβ with Rab11
Abstract
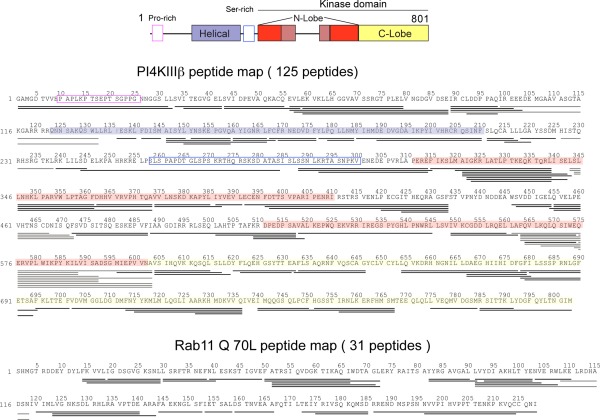
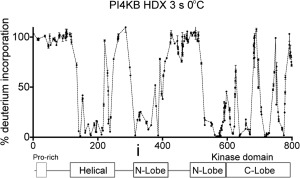
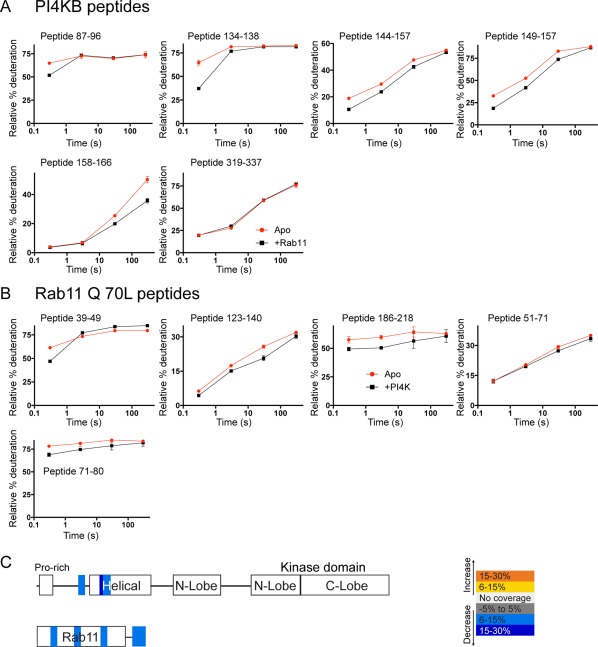
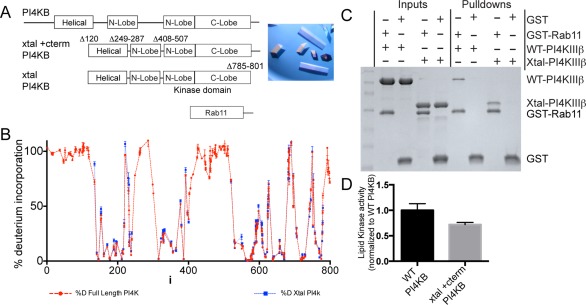
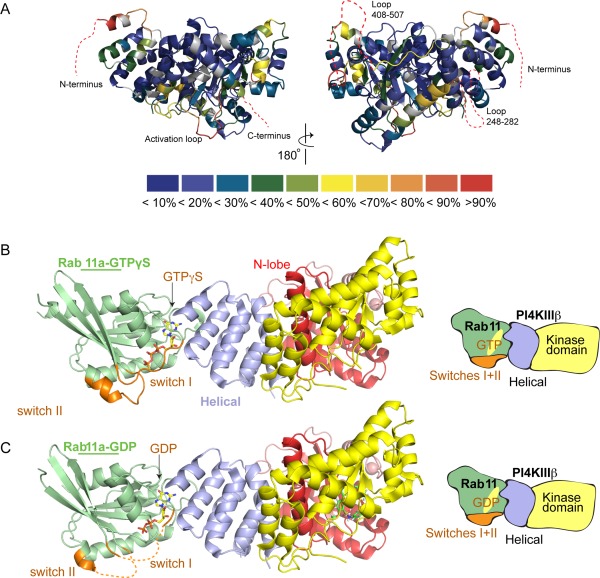
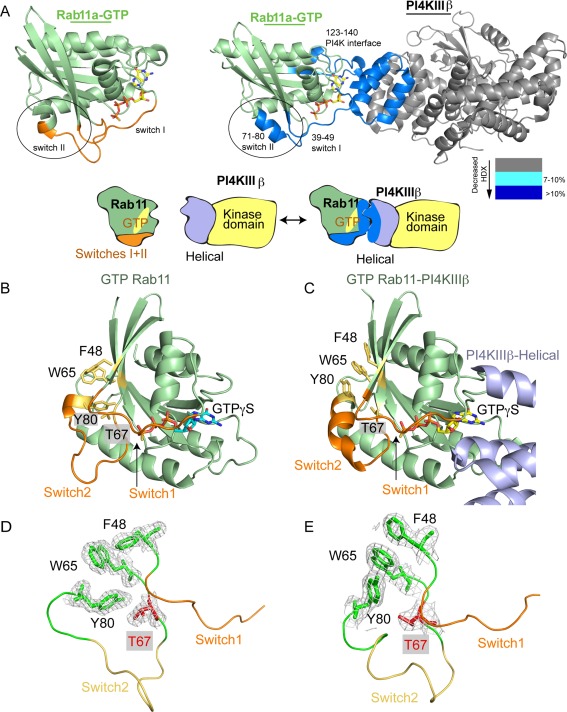
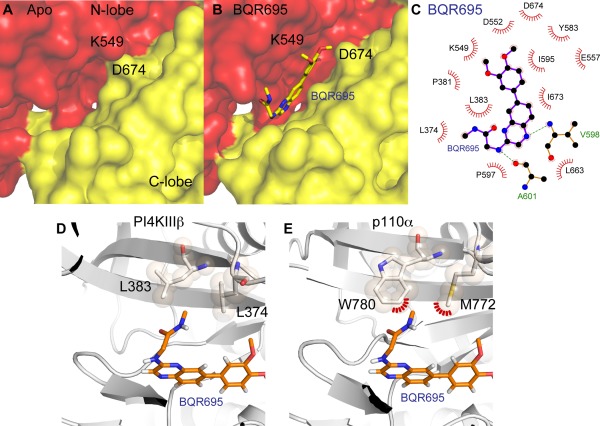
The ability of proteins to bind and interact with protein partners plays fundamental roles in many cellular contexts. X-ray crystallography has been a powerful approach to understand protein-protein interactions; however, a challenge in the crystallization of proteins and their complexes is the presence of intrinsically disordered regions. In this article, we describe an application of hydrogen deuterium exchange mass spectrometry (HDX-MS) to identify dynamic regions within type III phosphatidylinositol 4 kinase beta (PI4KIIIβ) in complex with the GTPase Rab11. This information was then used to design deletions that allowed for the production of diffraction quality crystals. Importantly, we also used HDX-MS to verify that the new construct was properly folded, consistent with it being catalytically and functionally active. Structures of PI4KIIIβ in an Apo state and bound to the potent inhibitor BQR695 in complex with both GTPγS and GDP loaded Rab11 were determined. This hybrid HDX-MS/crystallographic strategy revealed novel aspects of the PI4KIIIβ-Rab11 complex, as well as the molecular mechanism of potency of a PI4K specific inhibitor (BQR695). This approach is widely applicable to protein-protein complexes, and is an excellent strategy to optimize constructs for high-resolution structural approaches.
Keywords: HDX-MS; Rab11; X-ray crystallography; hydrogen deuterium exchange mass spectrometry; lipid signaling; phosphatidylinositol 4 kinase; phosphoinositide signaling; structural biology.
© 2016 The Protein Society.
Figures
References
-
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337:635–645. - PubMed
-
- Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB (2003) Protein disorder prediction: implications for structural proteomics. Struct Fold Des 11:1453–1459. - PubMed
-
- Tompa P (2012) Intrinsically disordered proteins: a 10‐year recap. Trends Biochem Sci 37:509–516. - PubMed
-
- Sugase K, Dyson HJ, Wright PE (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447:1021–1025. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
