

Functional Profiling of Unfamiliar Microbial Communities Using a Validated De Novo Assembly Metatranscriptome Pipeline
- PMID: 26756338
- PMCID: PMC4710500
- DOI: 10.1371/journal.pone.0146423
Functional Profiling of Unfamiliar Microbial Communities Using a Validated De Novo Assembly Metatranscriptome Pipeline
Abstract
Background: Metatranscriptomic landscapes can provide insights in functional relationships within natural microbial communities. Analysis of complex metatranscriptome datasets of these communities poses a considerable bioinformatic challenge since they are non-restricted with a varying number of participating strains and species. For RNA-Seq data a standard approach is to align the generated reads to a set of closely related reference genomes. This only works well for microbial communities for which a near complete catalogue of reference genomes is available at a small evolutionary distance. In this study, we focus on the design of a validated de novo metatranscriptome assembly pipeline for single-end Illumina RNA-Seq data to obtain functional and taxonomic profiles of murine microbial communities.
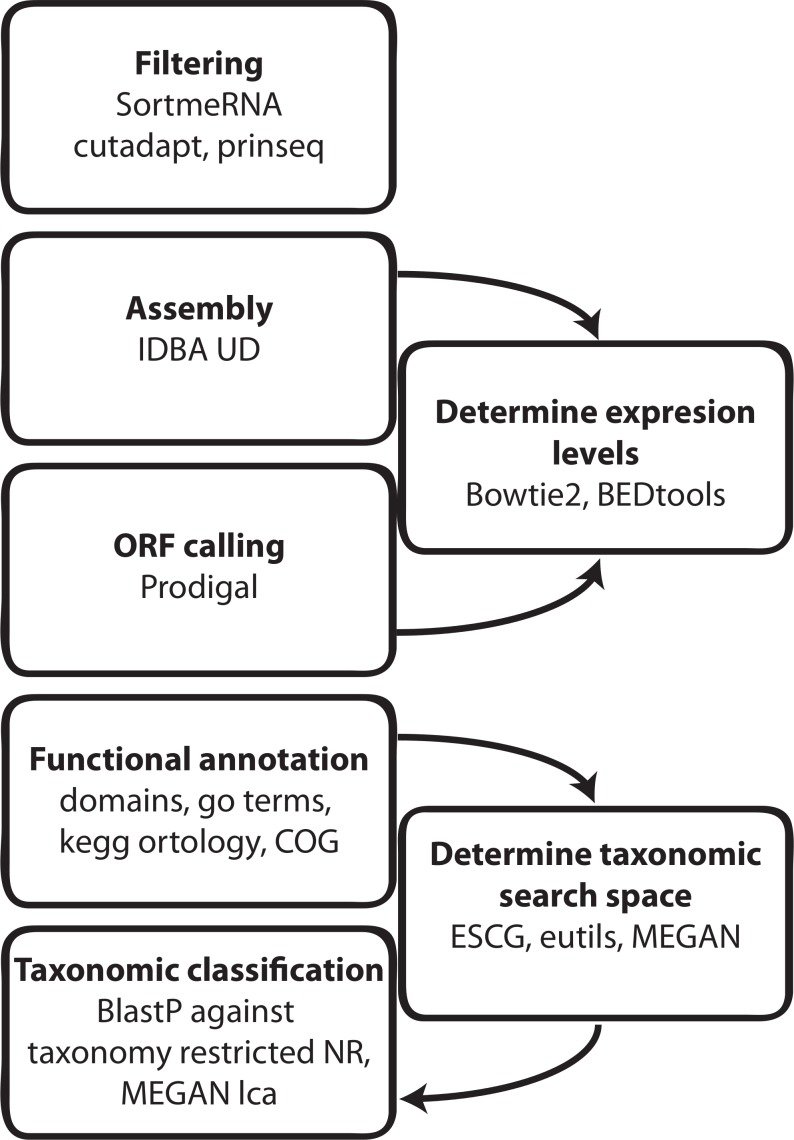
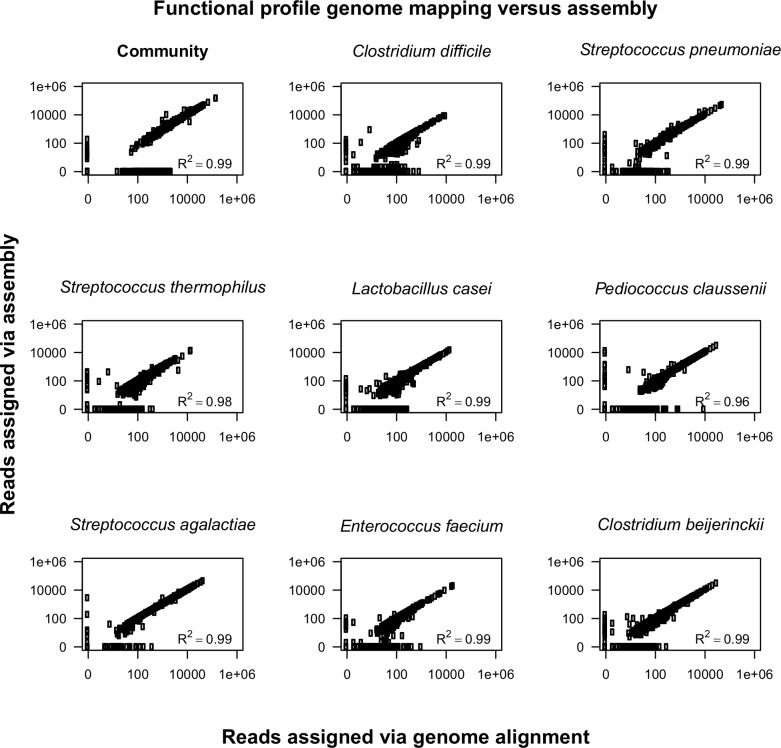
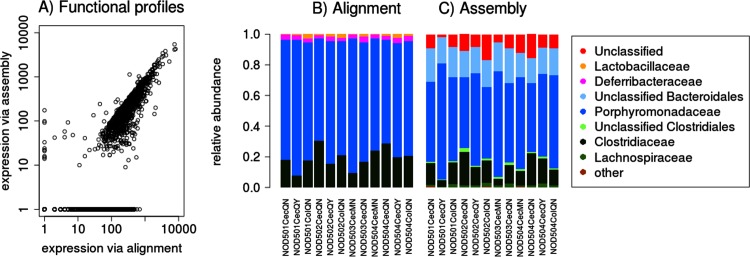
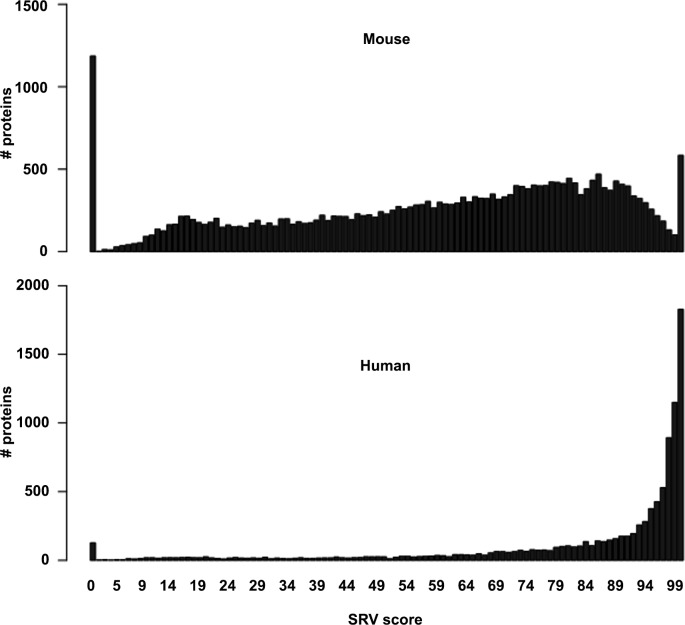
Results: The here developed de novo assembly metatranscriptome pipeline combined rRNA removal, IDBA-UD assembler, functional annotation and taxonomic classification. Different assemblers were tested and validated using RNA-Seq data from an in silico generated mock community and in vivo RNA-Seq data from a restricted microbial community taken from a mouse model colonized with Altered Schaedler Flora (ASF). Precision and recall of resulting gene expression, functional and taxonomic profiles were compared to those obtained with a standard alignment method. The validated pipeline was subsequently used to generate expression profiles from non-restricted cecal communities of four C57BL/6J mice fed on a high-fat high-protein diet spiked with an RNA-Seq data set from a well-characterized human sample. The spike in control was used to estimate precision and recall at assembly, functional and taxonomic level of non-restricted communities.
Conclusions: A generic de novo assembly pipeline for metatranscriptome data analysis was designed for microbial ecosystems, which can be applied for microbial metatranscriptome analysis in any chosen niche.
Conflict of interest statement
Figures
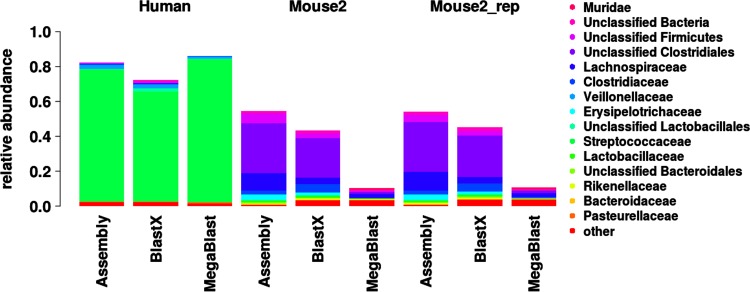
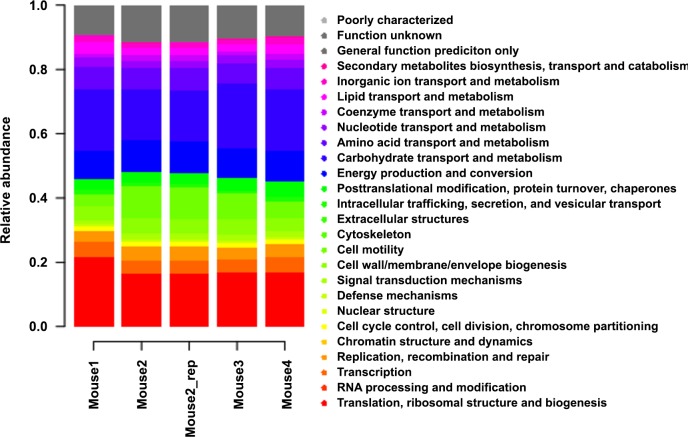
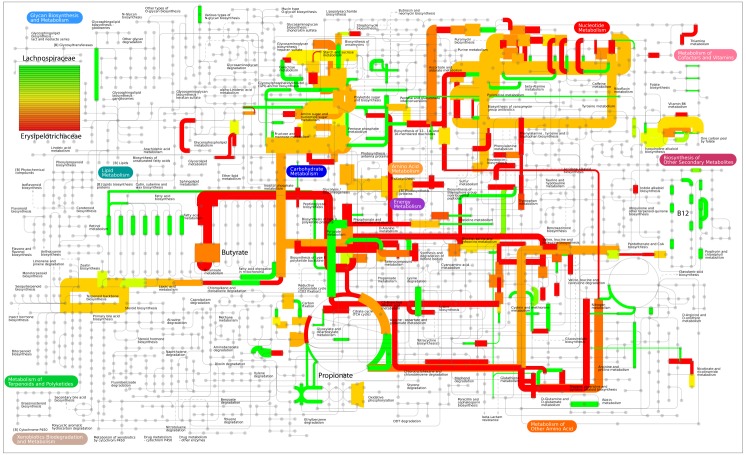
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
