

Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer's Disease
- PMID: 26758846
- PMCID: PMC4710776
- DOI: 10.1523/JNEUROSCI.2117-15.2016
Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer's Disease
Abstract
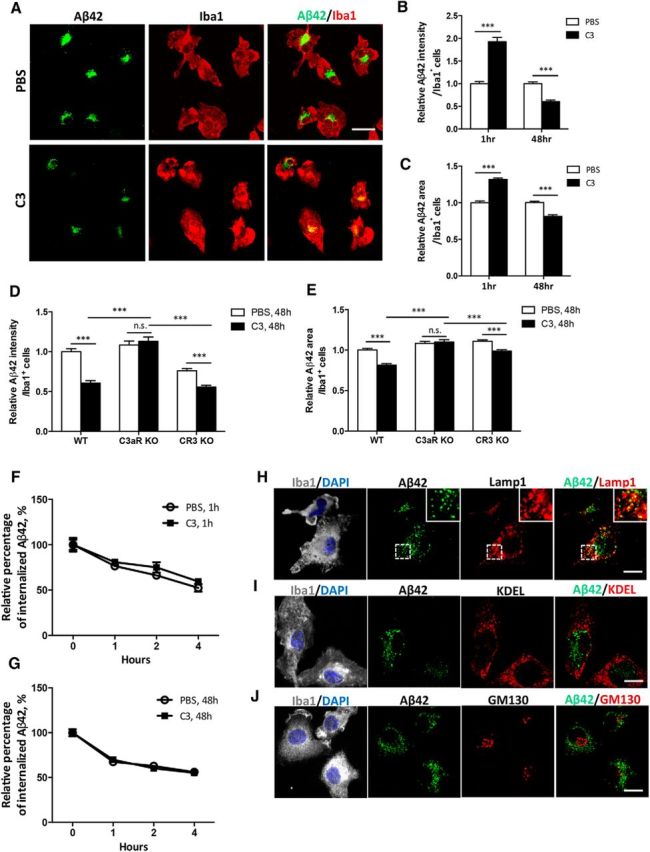
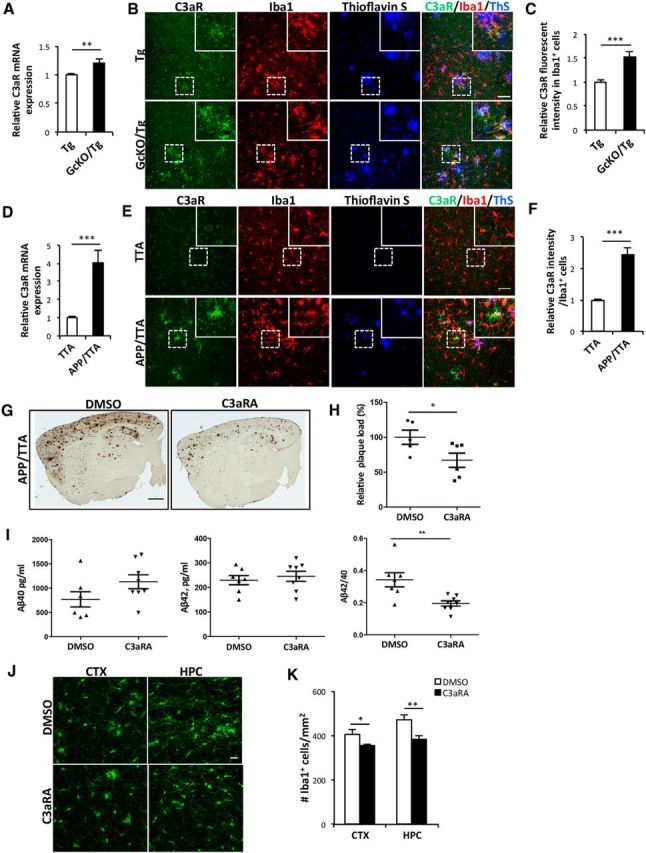
Increasing evidence supports a role of neuroinflammation in the pathogenesis of Alzheimer's disease (AD). Previously, we identified a neuron-glia signaling pathway whereby Aβ acts as an upstream activator of astroglial nuclear factor kappa B (NF-κB), leading to the release of complement C3, which acts on the neuronal C3a receptor (C3aR) to influence dendritic morphology and cognitive function. Here we report that astrocytic complement activation also regulates Aβ dynamics in vitro and amyloid pathology in AD mouse models through microglial C3aR. We show that in primary microglial cultures, acute C3 or C3a activation promotes, whereas chronic C3/C3a treatment attenuates, microglial phagocytosis and that the effect of chronic C3 exposure can be blocked by cotreatment with a C3aR antagonist and by genetic deletion of C3aR. We further demonstrate that Aβ pathology and neuroinflammation in amyloid precursor protein (APP) transgenic mice are worsened by astroglial NF-κB hyperactivation and resulting C3 elevation, whereas treatment with the C3aR antagonist (C3aRA) ameliorates plaque load and microgliosis. Our studies define a complement-dependent intercellular cross talk in which neuronal overproduction of Aβ activates astroglial NF-κB to elicit extracellular release of C3. This promotes a pathogenic cycle by which C3 in turn interacts with neuronal and microglial C3aR to alter cognitive function and impair Aβ phagocytosis. This feedforward loop can be effectively blocked by C3aR inhibition, supporting the therapeutic potential of C3aR antagonists under chronic neuroinflammation conditions.
Significance statement: The complement pathway is activated in Alzheimer's disease. Here we show that the central complement factor C3 secreted from astrocytes interacts with microglial C3a receptor (C3aR) to mediate β-amyloid pathology and neuroinflammation in AD mouse models. Our study provides support for targeting C3aR as a potential therapy for Alzheimer's disease.
Keywords: Alzheimer's disease; C3; C3a receptor; amyloid; mice; microglia.
Copyright © 2016 the authors 0270-6474/16/360577-13$15.00/0.
Figures
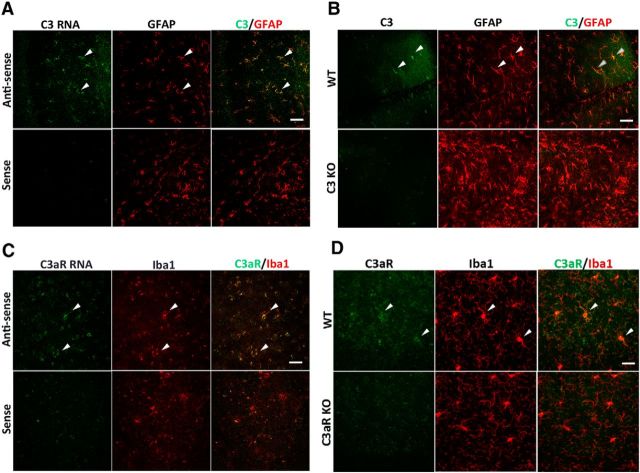
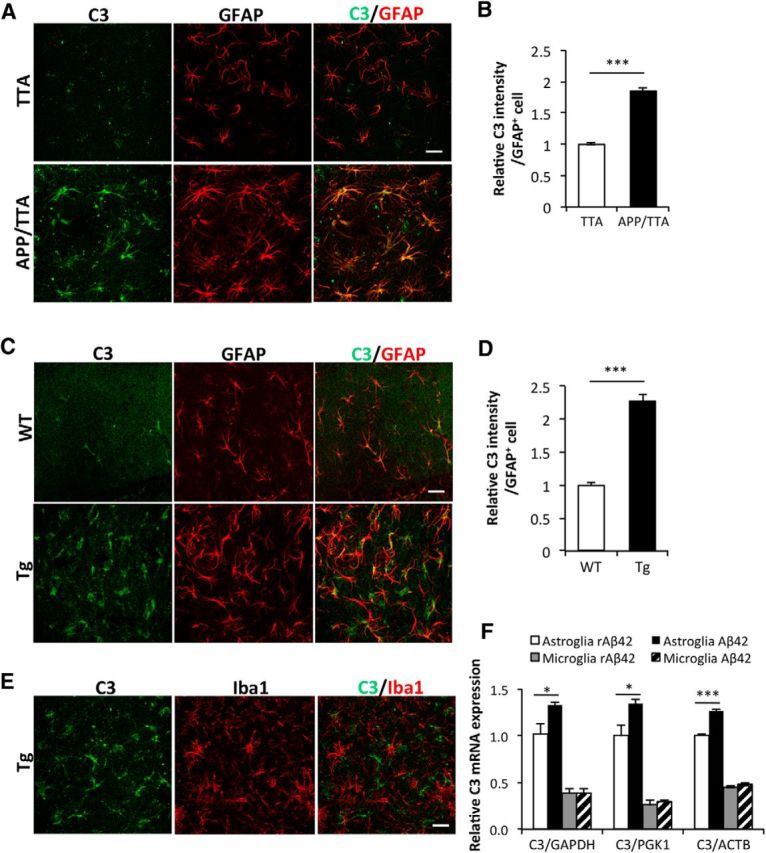
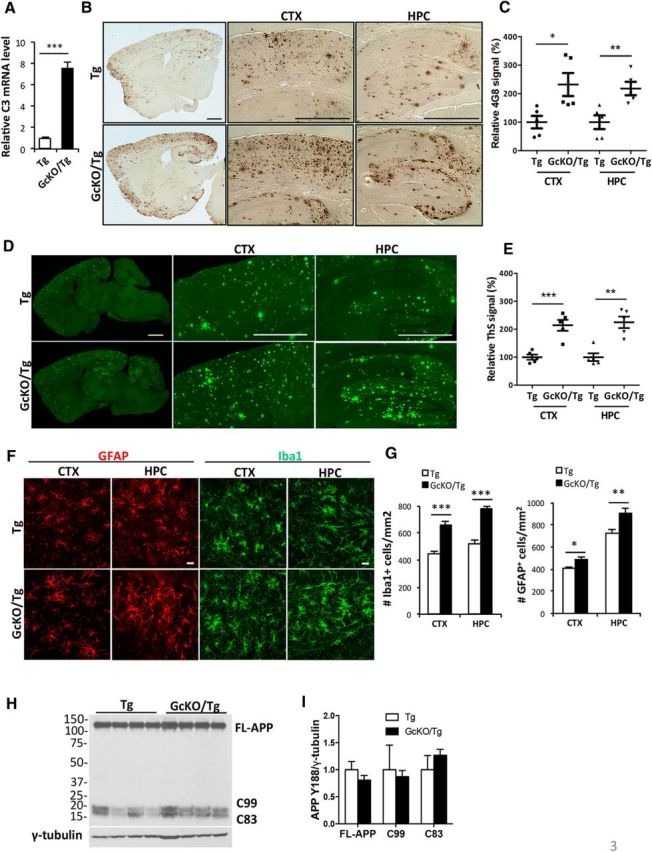
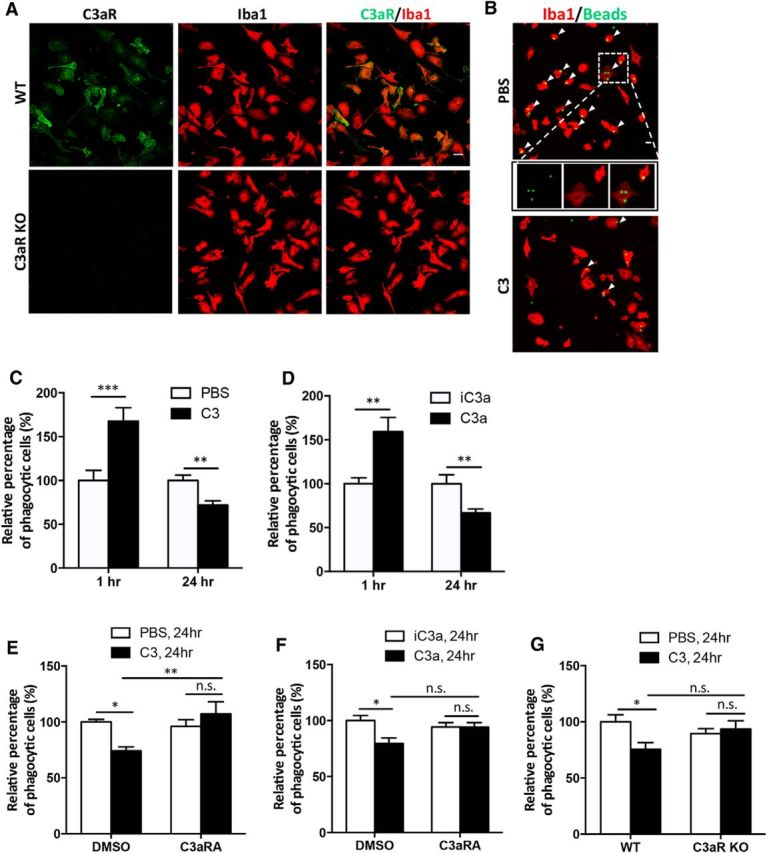
Similar articles
-
NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease.Neuron. 2015 Jan 7;85(1):101-115. doi: 10.1016/j.neuron.2014.11.018. Epub 2014 Dec 18. Neuron. 2015. PMID: 25533482 Free PMC article.
-
APP Regulates Microglial Phenotype in a Mouse Model of Alzheimer's Disease.J Neurosci. 2016 Aug 10;36(32):8471-86. doi: 10.1523/JNEUROSCI.4654-15.2016. J Neurosci. 2016. PMID: 27511018 Free PMC article.
-
Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer's disease.J Neurochem. 2008 Sep;106(5):2080-92. doi: 10.1111/j.1471-4159.2008.05558.x. Epub 2008 Jul 9. J Neurochem. 2008. PMID: 18624920 Free PMC article.
-
Refining the interactions between microglia and astrocytes in Alzheimer's disease pathology.Neuroscience. 2025 May 7;573:183-197. doi: 10.1016/j.neuroscience.2025.03.033. Epub 2025 Mar 20. Neuroscience. 2025. PMID: 40120713 Review.
-
Complement 3a induces the synapse loss via C3aR in mitochondria-dependent NLRP3 activating mechanisms during the development and progression of Alzheimer's disease.Neurosci Biobehav Rev. 2024 Oct;165:105868. doi: 10.1016/j.neubiorev.2024.105868. Epub 2024 Aug 30. Neurosci Biobehav Rev. 2024. PMID: 39218048 Review.
Cited by
-
Interaction between autophagy and the NLRP3 inflammasome in Alzheimer's and Parkinson's disease.Front Aging Neurosci. 2022 Oct 3;14:1018848. doi: 10.3389/fnagi.2022.1018848. eCollection 2022. Front Aging Neurosci. 2022. PMID: 36262883 Free PMC article. Review.
-
Affective Immunology: The Crosstalk Between Microglia and Astrocytes Plays Key Role?Front Immunol. 2020 Aug 20;11:1818. doi: 10.3389/fimmu.2020.01818. eCollection 2020. Front Immunol. 2020. PMID: 32973758 Free PMC article. Review.
-
NF-κB Signaling in Astrocytes Modulates Brain Inflammation and Neuronal Injury Following Sequential Exposure to Manganese and MPTP During Development and Aging.Toxicol Sci. 2020 Oct 1;177(2):506-520. doi: 10.1093/toxsci/kfaa115. Toxicol Sci. 2020. PMID: 32692843 Free PMC article.
-
Rheb-mTOR activation rescues Aβ-induced cognitive impairment and memory function by restoring miR-146 activity in glial cells.Mol Ther Nucleic Acids. 2021 Apr 24;24:868-887. doi: 10.1016/j.omtn.2021.04.008. eCollection 2021 Jun 4. Mol Ther Nucleic Acids. 2021. PMID: 34094708 Free PMC article.
-
Increased Type I interferon signaling and brain endothelial barrier dysfunction in an experimental model of Alzheimer's disease.Sci Rep. 2022 Oct 1;12(1):16488. doi: 10.1038/s41598-022-20889-y. Sci Rep. 2022. PMID: 36182964 Free PMC article.
References
-
- Ames RS, Lee D, Foley JJ, Jurewicz AJ, Tornetta MA, Bautsch W, Settmacher B, Klos A, Erhard KF, Cousins RD, Sulpizio AC, Hieble JP, McCafferty G, Ward KW, Adams JL, Bondinell WE, Underwood DC, Osborn RR, Badger AM, Sarau HM. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol. 2001;166:6341–6348. doi: 10.4049/jimmunol.166.10.6341. - DOI - PubMed
-
- Banda NK, Hyatt S, Antonioli AH, White JT, Glogowska M, Takahashi K, Merkel TJ, Stahl GL, Mueller-Ortiz S, Wetsel R, Arend WP, Holers VM. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. J Immunol. 2012;188:1469–1478. doi: 10.4049/jimmunol.1102310. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous