

Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline
- PMID: 26760044
- PMCID: PMC4880116
- DOI: 10.1210/jc.2015-1710
Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline
Abstract
Objective: This clinical practice guideline addresses the diagnosis and treatment of primary adrenal insufficiency.
Participants: The Task Force included a chair, selected by The Clinical Guidelines Subcommittee of the Endocrine Society, eight additional clinicians experienced with the disease, a methodologist, and a medical writer. The co-sponsoring associations (European Society of Endocrinology and the American Association for Clinical Chemistry) had participating members. The Task Force received no corporate funding or remuneration in connection with this review.
Evidence: This evidence-based guideline was developed using the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) system to determine the strength of recommendations and the quality of evidence.
Consensus process: The evidence used to formulate recommendations was derived from two commissioned systematic reviews as well as other published systematic reviews and studies identified by the Task Force. The guideline was reviewed and approved sequentially by the Endocrine Society's Clinical Guidelines Subcommittee and Clinical Affairs Core Committee, members responding to a web posting, and the Endocrine Society Council. At each stage, the Task Force incorporated changes in response to written comments.
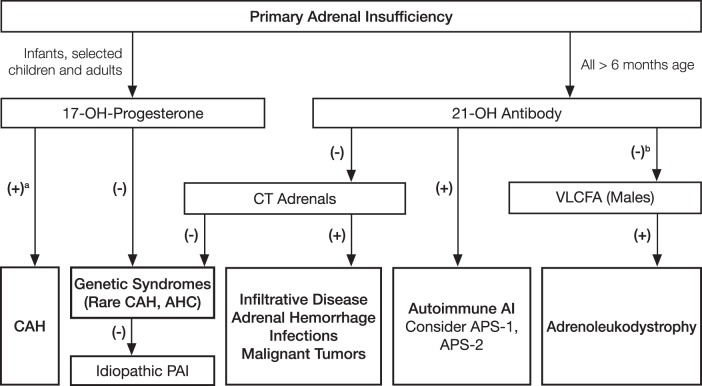
Conclusions: We recommend diagnostic tests for the exclusion of primary adrenal insufficiency in all patients with indicative clinical symptoms or signs. In particular, we suggest a low diagnostic (and therapeutic) threshold in acutely ill patients, as well as in patients with predisposing factors. This is also recommended for pregnant women with unexplained persistent nausea, fatigue, and hypotension. We recommend a short corticotropin test (250 μg) as the "gold standard" diagnostic tool to establish the diagnosis. If a short corticotropin test is not possible in the first instance, we recommend an initial screening procedure comprising the measurement of morning plasma ACTH and cortisol levels. Diagnosis of the underlying cause should include a validated assay of autoantibodies against 21-hydroxylase. In autoantibody-negative individuals, other causes should be sought. We recommend once-daily fludrocortisone (median, 0.1 mg) and hydrocortisone (15-25 mg/d) or cortisone acetate replacement (20-35 mg/d) applied in two to three daily doses in adults. In children, hydrocortisone (∼8 mg/m(2)/d) is recommended. Patients should be educated about stress dosing and equipped with a steroid card and glucocorticoid preparation for parenteral emergency administration. Follow-up should aim at monitoring appropriate dosing of corticosteroids and associated autoimmune diseases, particularly autoimmune thyroid disease.
Figures
References
-
- Addison T. On the Constitutional and Local Effects of Disease of the Supra-renal Capsules. London, UK: Samuel Highley; 1855.
-
- Bleicken B, Hahner S, Ventz M, Quinkler M. Delayed diagnosis of adrenal insufficiency is common: a cross-sectional study in 216 patients. Am J Med Sci. 2010;339:525–531. - PubMed
-
- Løvås K, Loge JH, Husebye ES. Subjective health status in Norwegian patients with Addison's disease. Clin Endocrinol (Oxf). 2002;56:581–588. - PubMed
-
- Hahner S, Allolio B. Therapeutic management of adrenal insufficiency. Best Pract Res Clin Endocrinol Metab. 2009;23:167–179. - PubMed
-
- Reisch N, Arlt W. Fine tuning for quality of life: 21st century approach to treatment of Addison's disease. Endocrinol Metab Clin North Am. 2009;38:407–418, ix–x. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
