

Divergent clonal selection dominates medulloblastoma at recurrence
- PMID: 26760213
- PMCID: PMC4936195
- DOI: 10.1038/nature16478
Divergent clonal selection dominates medulloblastoma at recurrence
Abstract
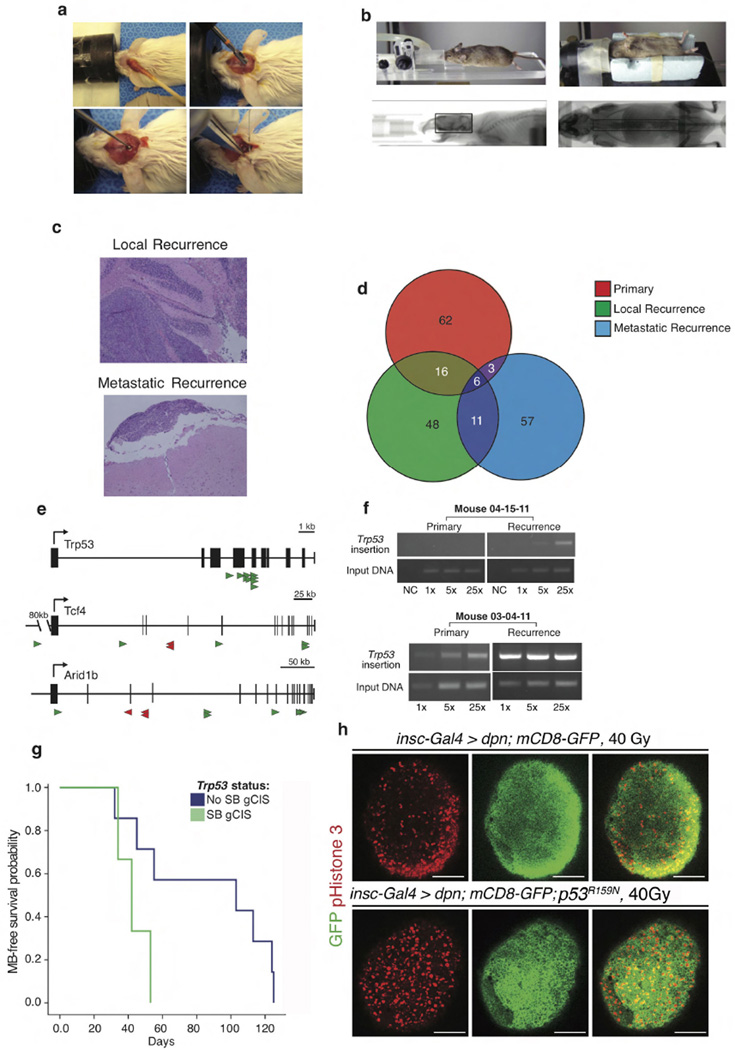
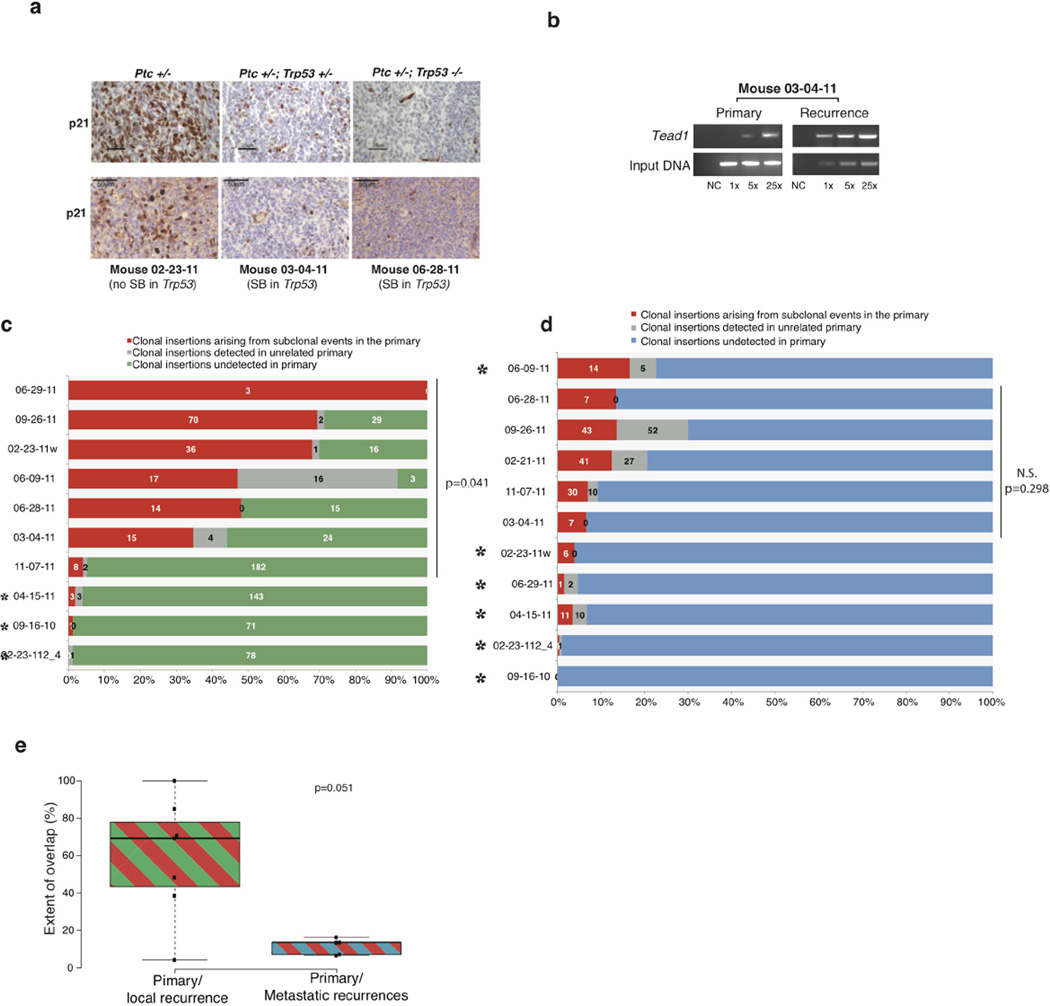
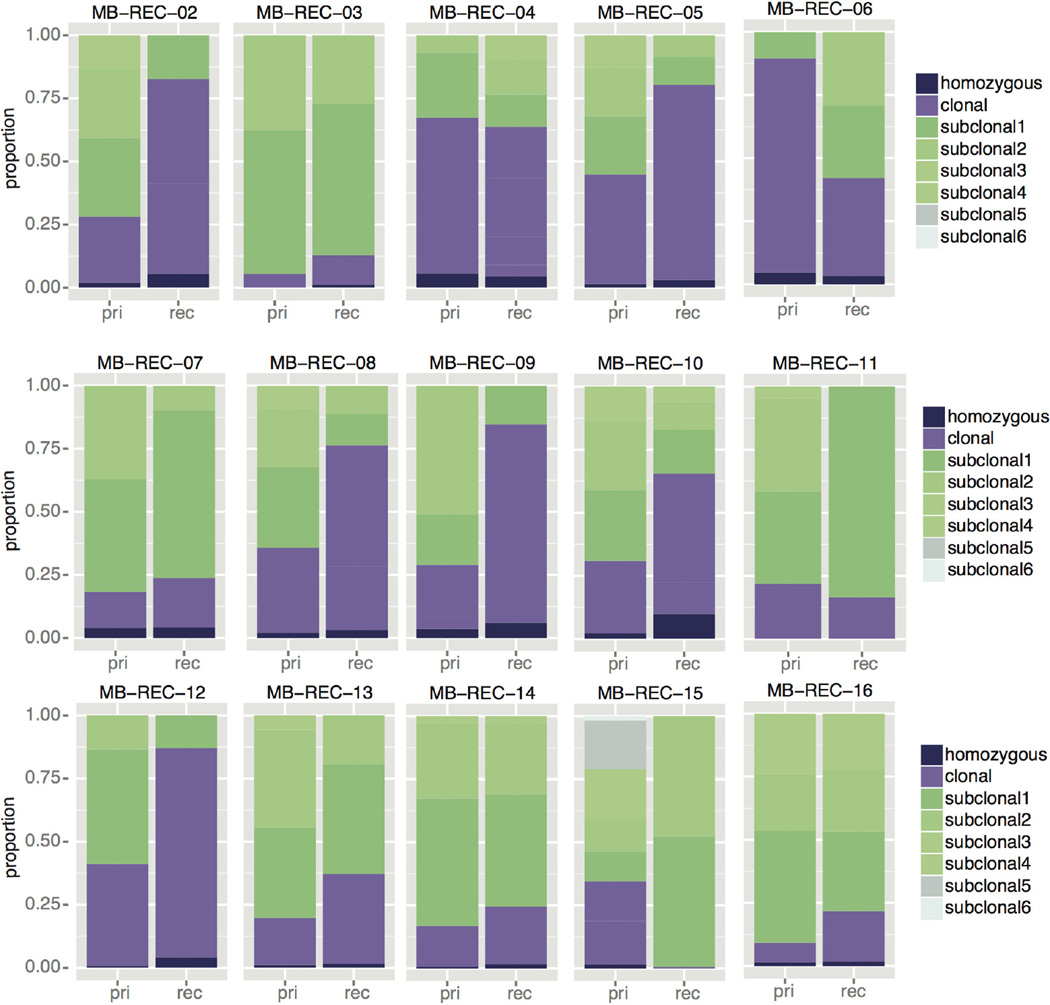
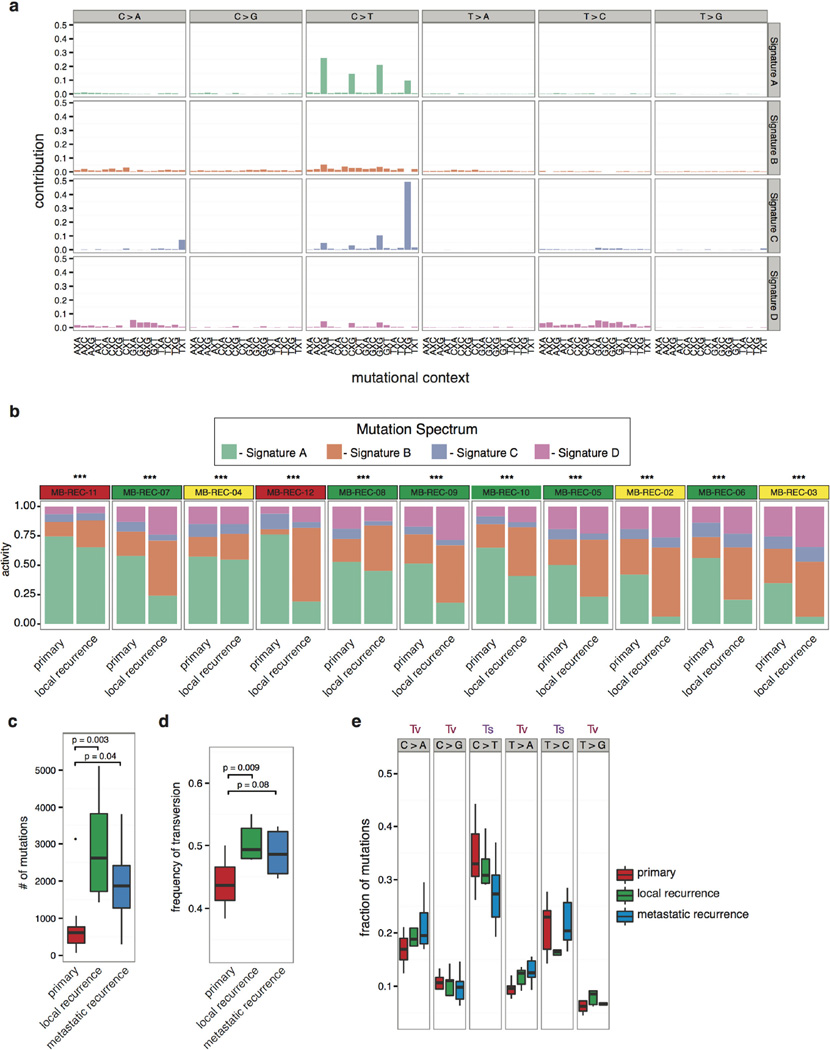
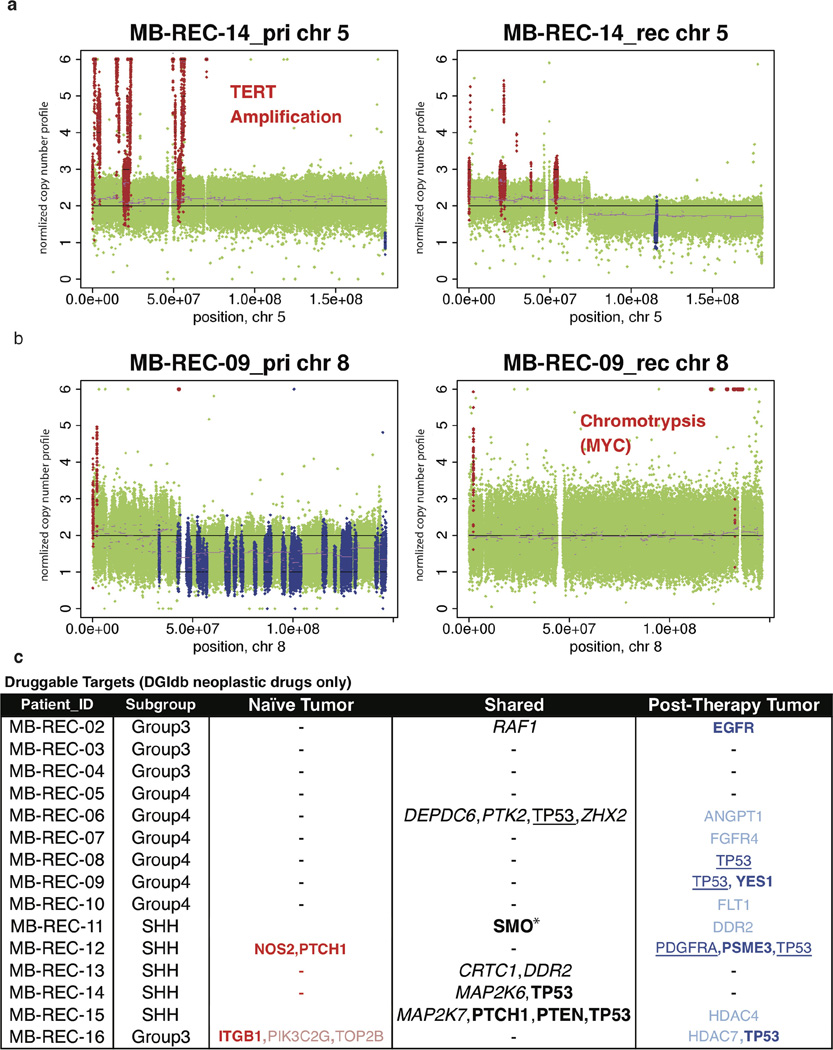
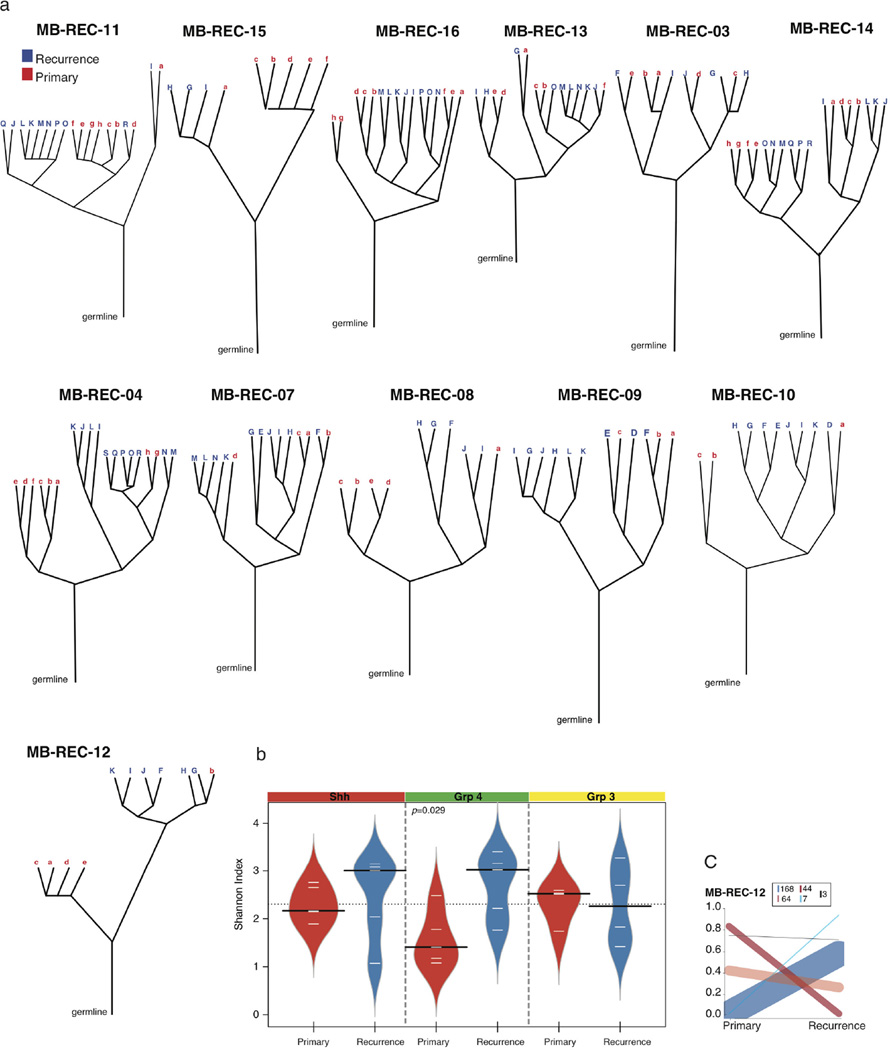
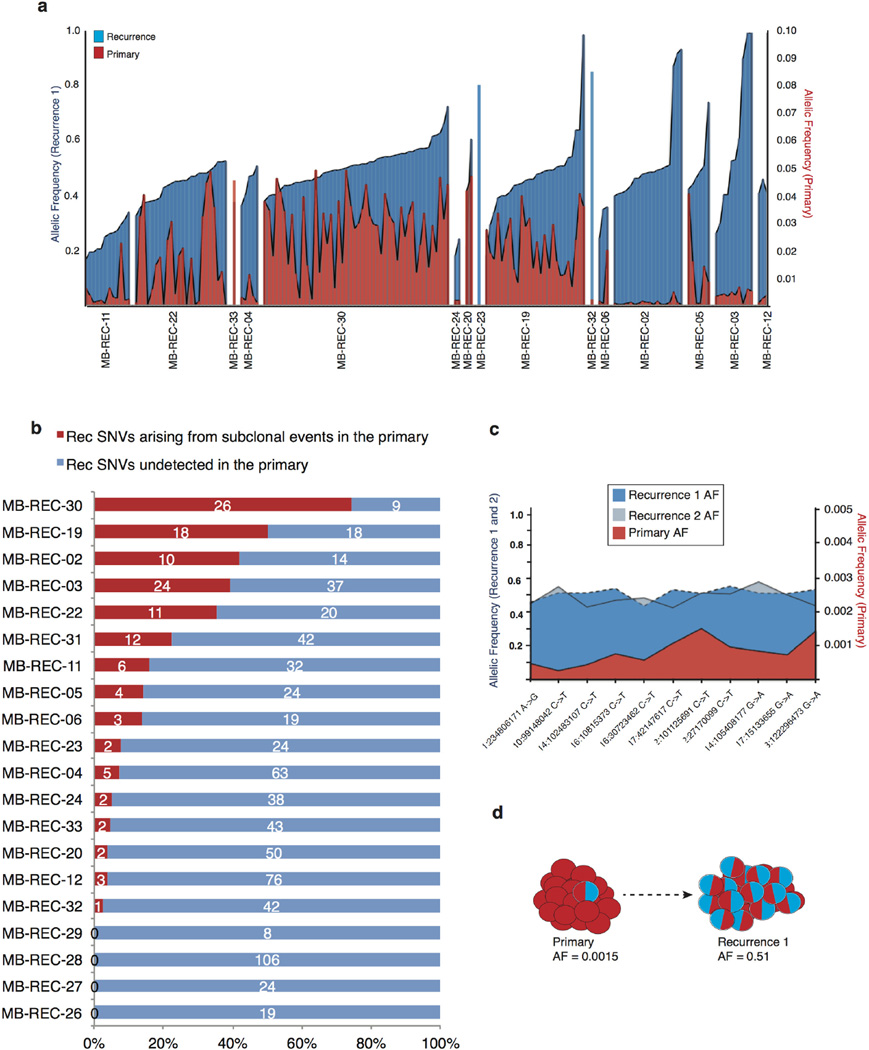
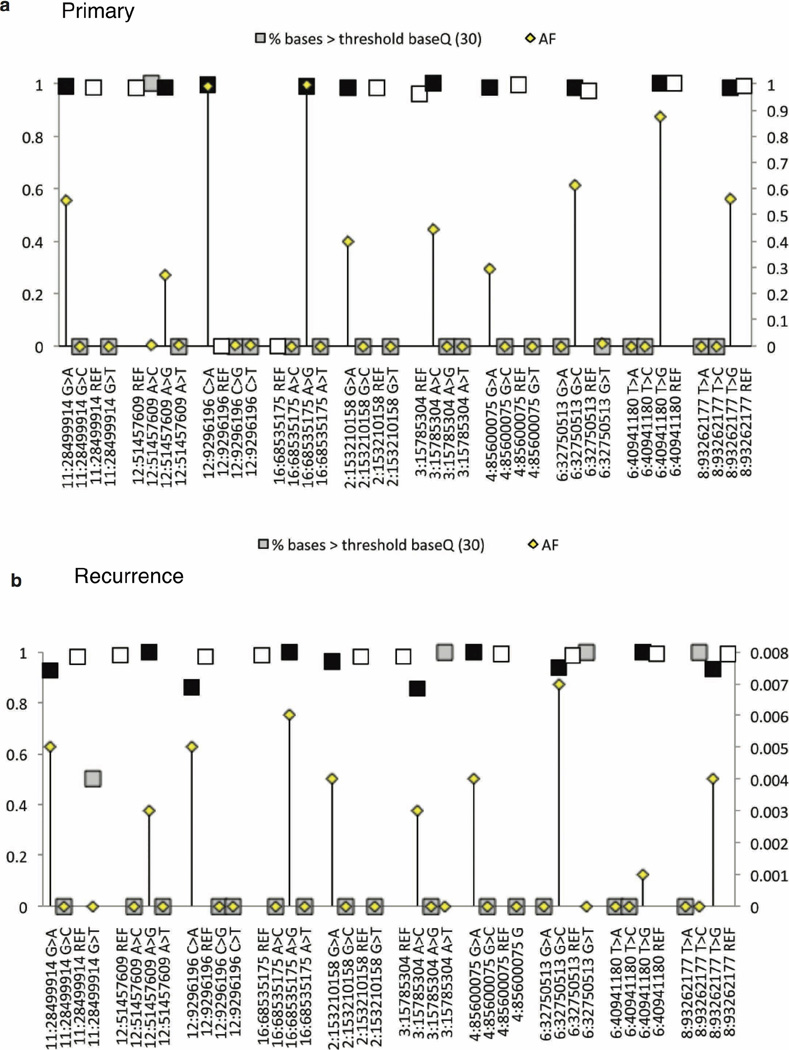
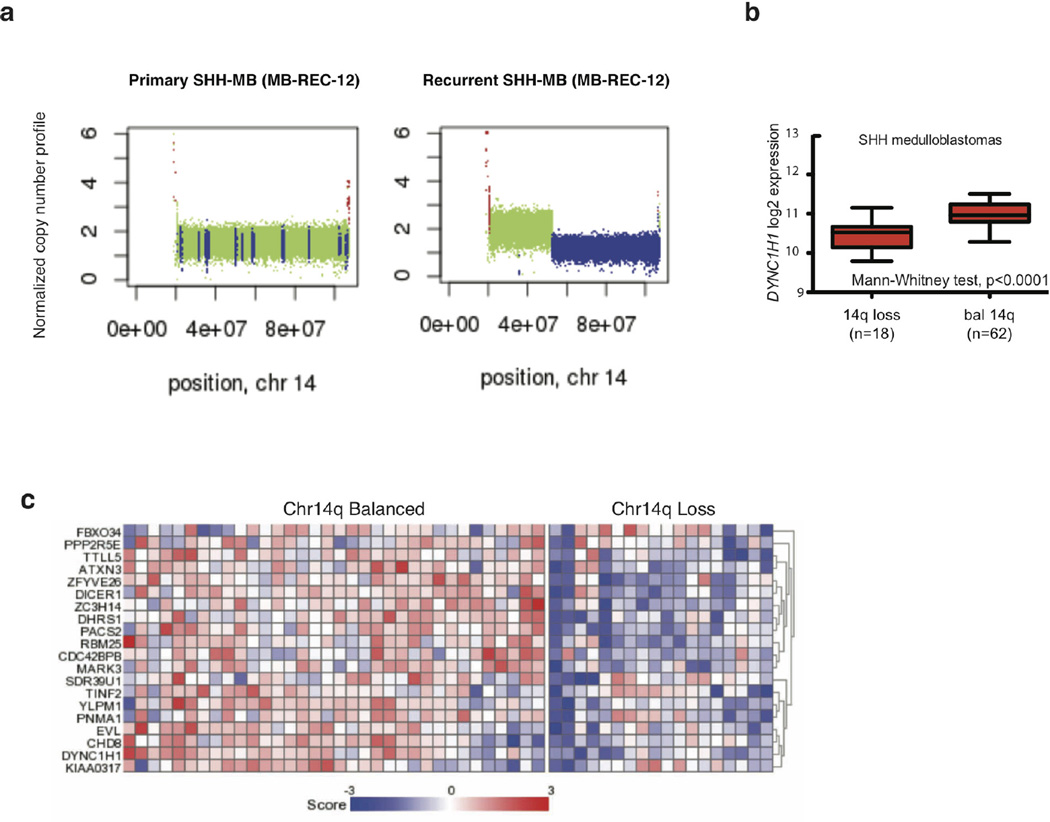
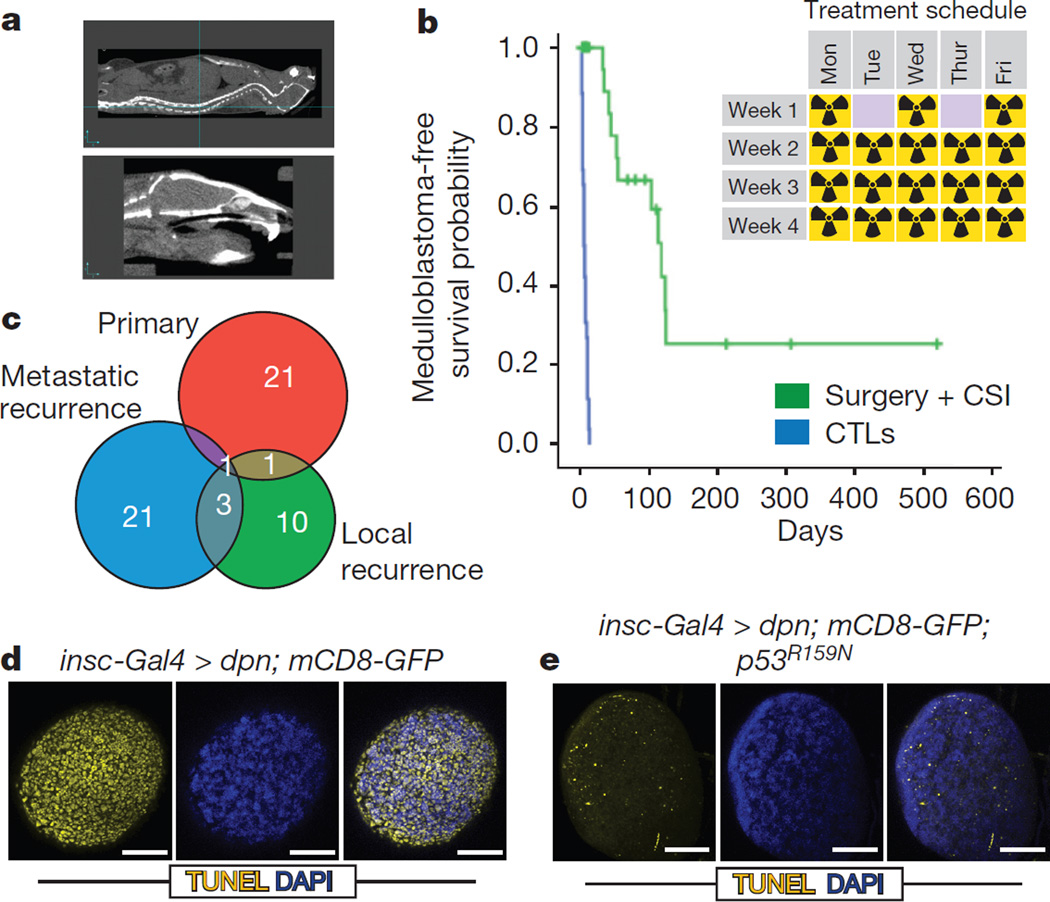
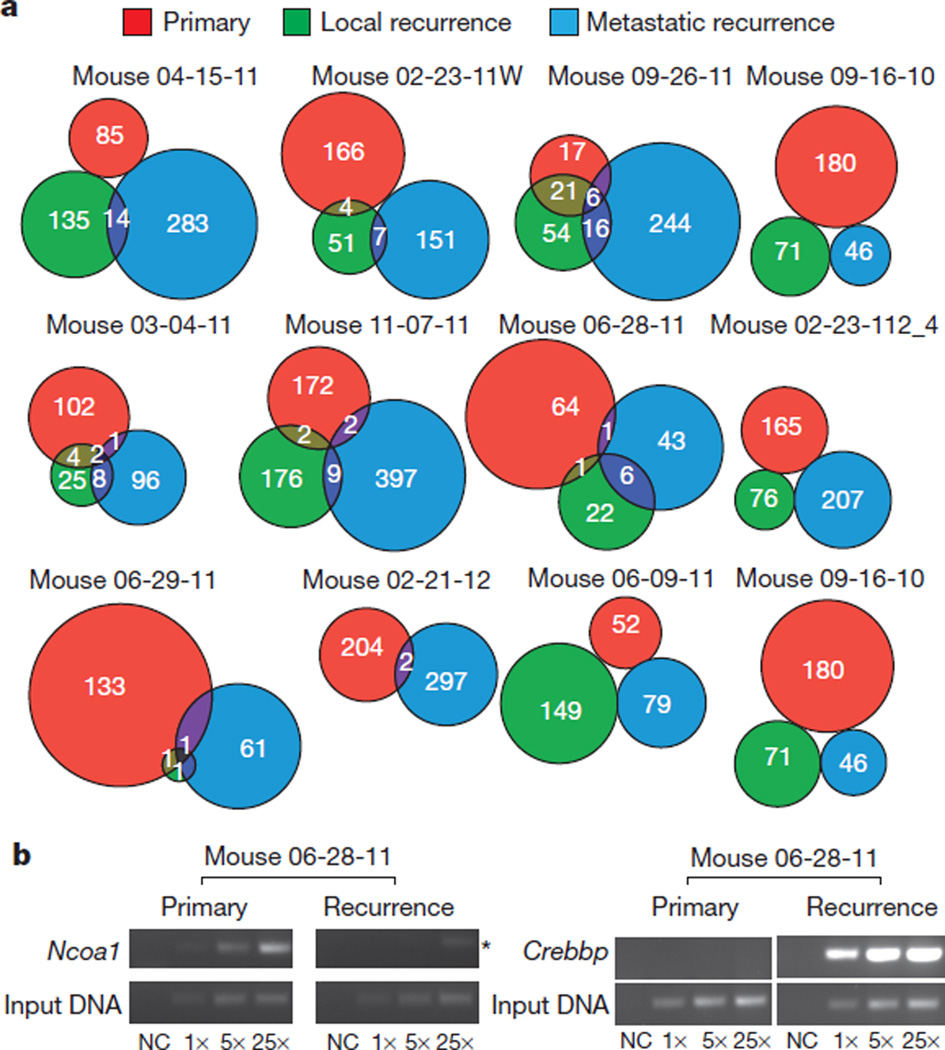
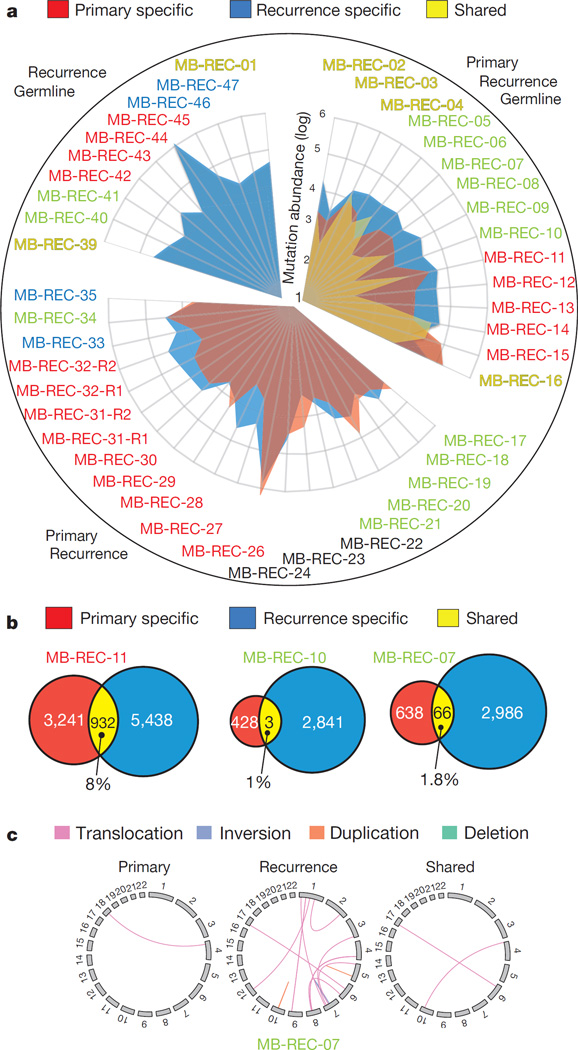
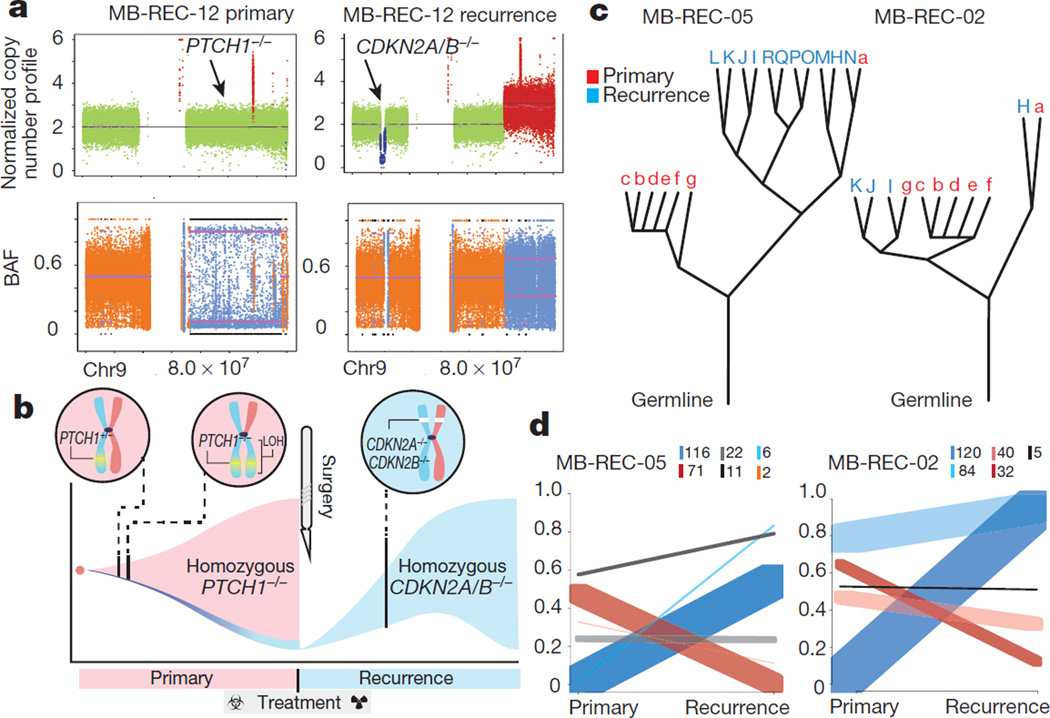
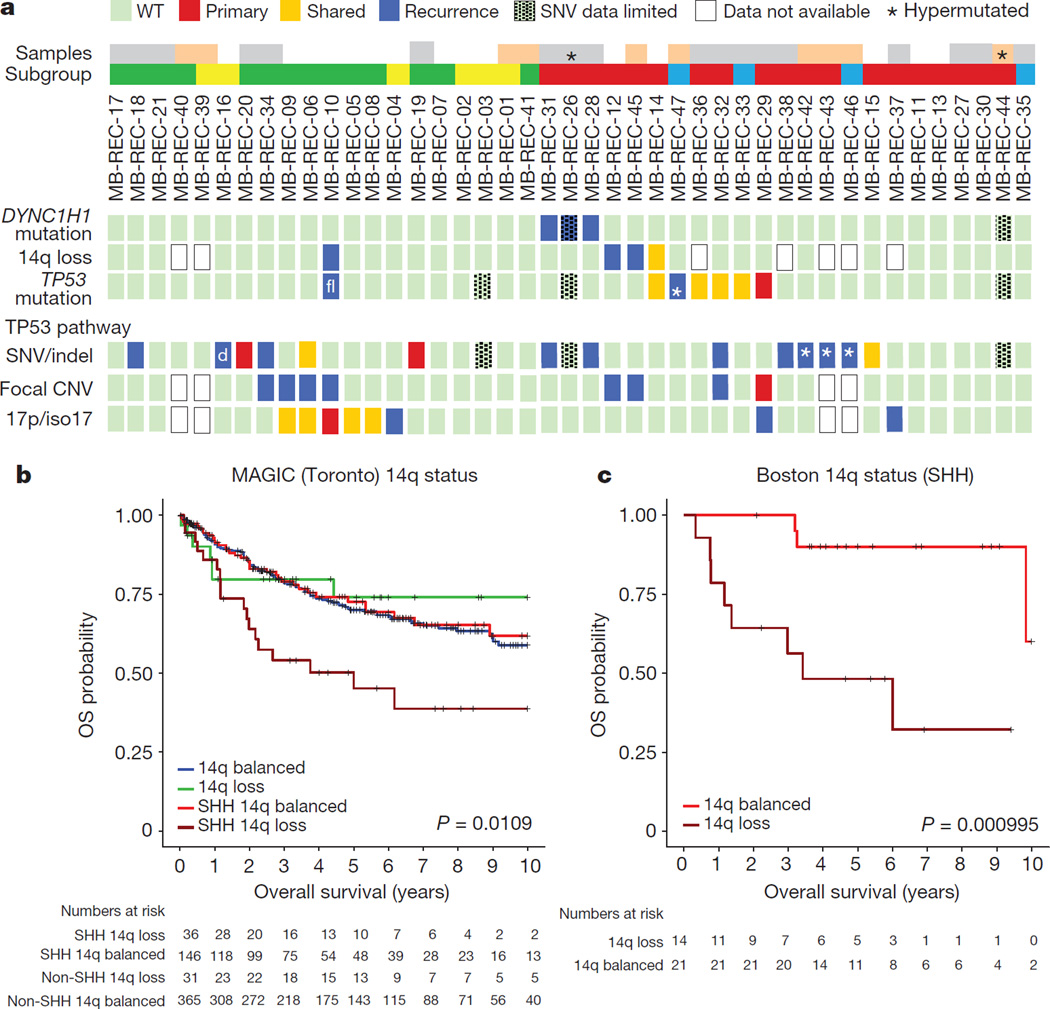
The development of targeted anti-cancer therapies through the study of cancer genomes is intended to increase survival rates and decrease treatment-related toxicity. We treated a transposon-driven, functional genomic mouse model of medulloblastoma with 'humanized' in vivo therapy (microneurosurgical tumour resection followed by multi-fractionated, image-guided radiotherapy). Genetic events in recurrent murine medulloblastoma exhibit a very poor overlap with those in matched murine diagnostic samples (<5%). Whole-genome sequencing of 33 pairs of human diagnostic and post-therapy medulloblastomas demonstrated substantial genetic divergence of the dominant clone after therapy (<12% diagnostic events were retained at recurrence). In both mice and humans, the dominant clone at recurrence arose through clonal selection of a pre-existing minor clone present at diagnosis. Targeted therapy is unlikely to be effective in the absence of the target, therefore our results offer a simple, proximal, and remediable explanation for the failure of prior clinical trials of targeted therapy.
Figures

Comment in
-
Genetics: Divergence shapes recurrence.Nat Rev Clin Oncol. 2016 Mar;13(3):136. doi: 10.1038/nrclinonc.2016.14. Epub 2016 Feb 2. Nat Rev Clin Oncol. 2016. PMID: 26831182 No abstract available.
References
-
- Hovestadt V, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–541. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
