

Acute Kidney Injury
- PMID: 26768243
- PMCID: PMC4845743
- DOI: 10.1146/annurev-med-050214-013407
Acute Kidney Injury
Abstract
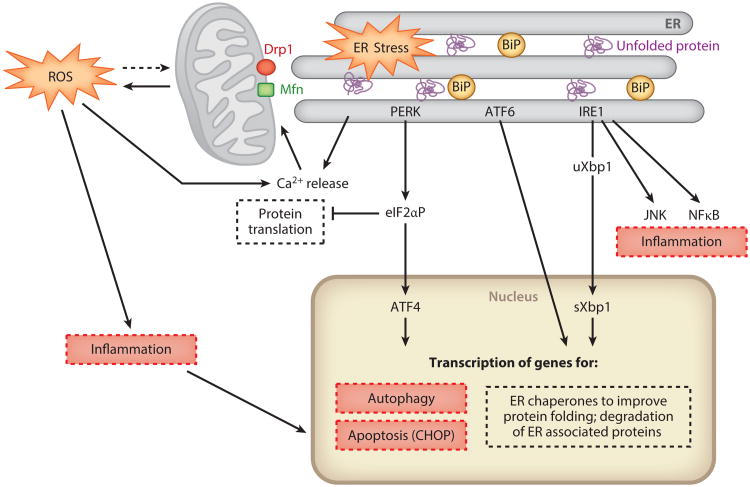
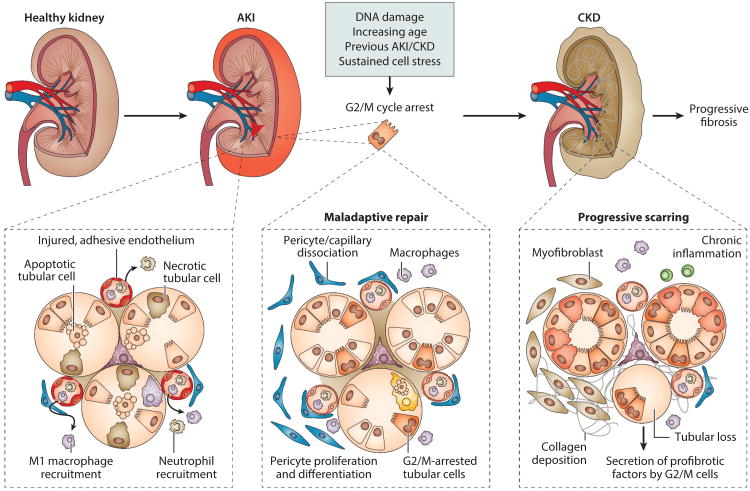
Acute kidney injury (AKI) is a global public health concern associated with high morbidity, mortality, and healthcare costs. Other than dialysis, no therapeutic interventions reliably improve survival, limit injury, or speed recovery. Despite recognized shortcomings of in vivo animal models, the underlying pathophysiology of AKI and its consequence, chronic kidney disease (CKD), is rich with biological targets. We review recent findings relating to the renal vasculature and cellular stress responses, primarily the intersection of the unfolded protein response, mitochondrial dysfunction, autophagy, and the innate immune response. Maladaptive repair mechanisms that persist following the acute phase promote inflammation and fibrosis in the chronic phase. Here macrophages, growth-arrested tubular epithelial cells, the endothelium, and surrounding pericytes are key players in the progression to chronic disease. Better understanding of these complex interacting pathophysiological mechanisms, their relative importance in humans, and the utility of biomarkers will lead to therapeutic strategies to prevent and treat AKI or impede progression to CKD or end-stage renal disease (ESRD).
Keywords: biomarkers; chronic kidney disease progression; maladaptive repair; nephrotoxicity; pathophysiology; renal ischemia-reperfusion.
Figures
References
-
- Mehta RL, Cerda J, Burdmann EA, et al. International Society of Nephrology's 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. 2015;385:1–28. - PubMed
-
- Yang L, Xing G, Wang L, et al. Acute kidney injury in China: a cross-sectional survey. Lancet. 2015;386:1465–71. - PubMed
-
- Cerda J, Bagga A, Kher V, Chakravarthi RM. The contrasting characteristics of acute kidney injury in developed and developing countries. Nat Clin Pract Nephrol. 2008;4:138–53. - PubMed
-
- Jha V, Parameswaran S. Community-acquired acute kidney injury in tropical countries. Nat Rev Nephrol. 2013;9:278–90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
