

Network-Thinking: Graphs to Analyze Microbial Complexity and Evolution
- PMID: 26774999
- PMCID: PMC4766943
- DOI: 10.1016/j.tim.2015.12.003
Network-Thinking: Graphs to Analyze Microbial Complexity and Evolution
Abstract
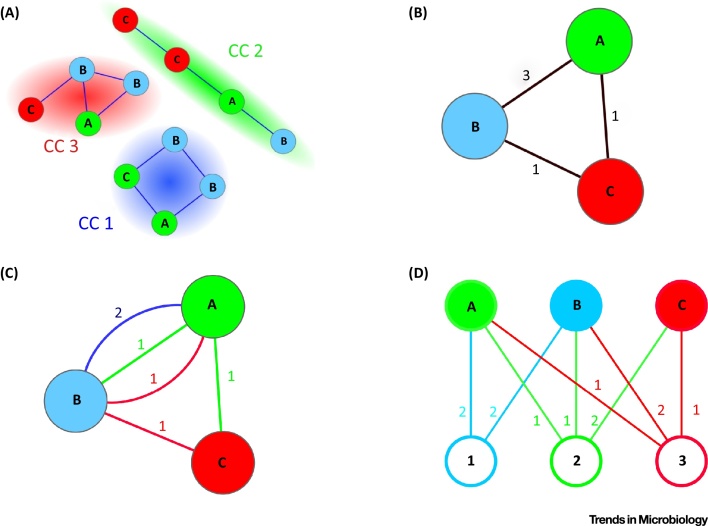
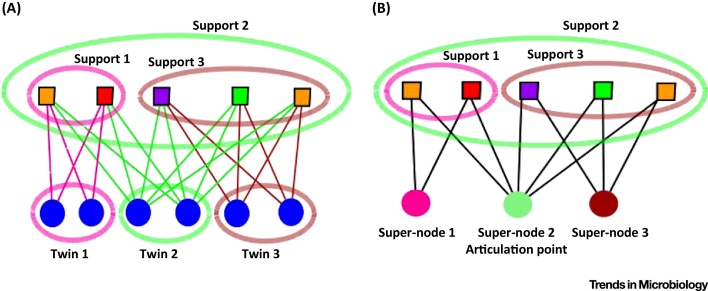
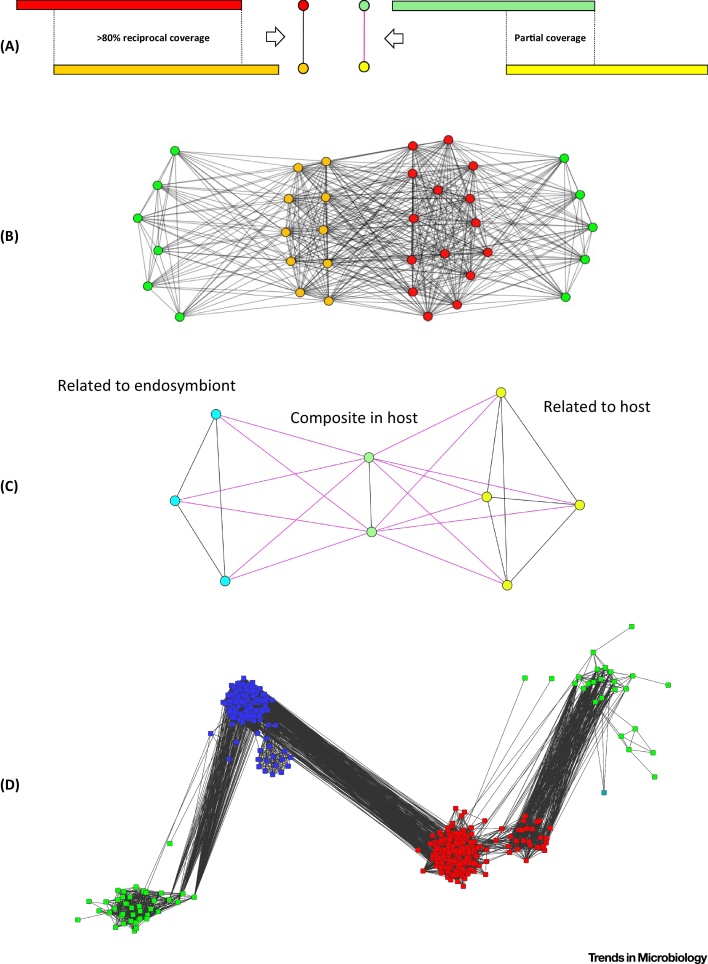
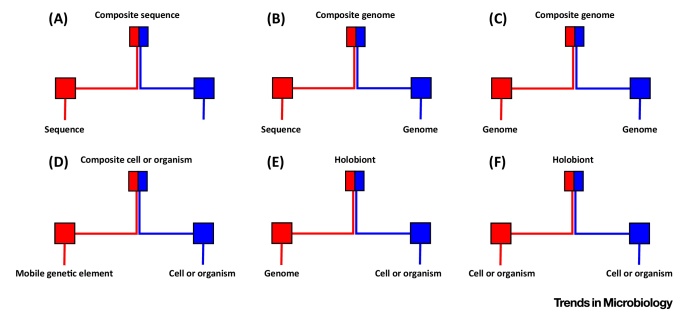
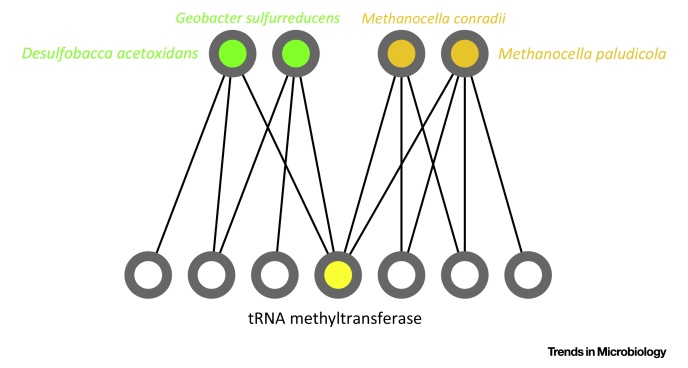
The tree model and tree-based methods have played a major, fruitful role in evolutionary studies. However, with the increasing realization of the quantitative and qualitative importance of reticulate evolutionary processes, affecting all levels of biological organization, complementary network-based models and methods are now flourishing, inviting evolutionary biology to experience a network-thinking era. We show how relatively recent comers in this field of study, that is, sequence-similarity networks, genome networks, and gene families-genomes bipartite graphs, already allow for a significantly enhanced usage of molecular datasets in comparative studies. Analyses of these networks provide tools for tackling a multitude of complex phenomena, including the evolution of gene transfer, composite genes and genomes, evolutionary transitions, and holobionts.
Keywords: bipartite graph; evolution; gene transfer; graph theory; introgression; symbiosis.
Copyright © 2016 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
References
-
- Darwin C. John Murray; 1859. On the Origin of Species by Means of Natural Selection.
-
- O’Hara R.J. Population thinking and tree thinking in systematics. Zool. Scr. 1997;26:323–329.
-
- Doolittle W.F. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2129. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
