

Old versus New Mechanisms in the Pathogenesis of ALS
- PMID: 26779612
- PMCID: PMC8029354
- DOI: 10.1111/bpa.12355
Old versus New Mechanisms in the Pathogenesis of ALS
Abstract
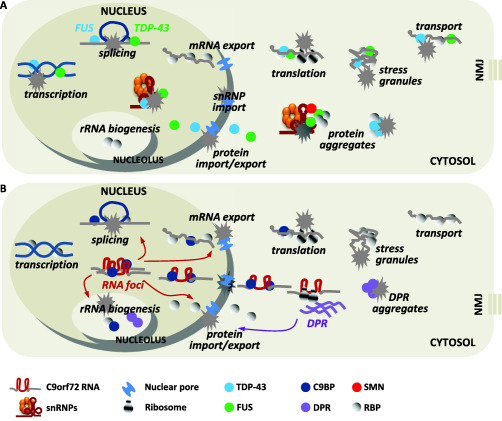
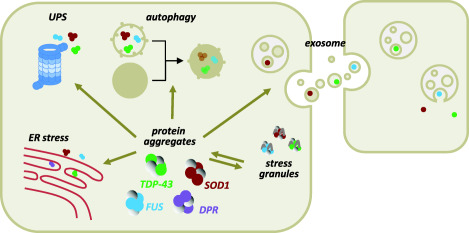
Amyotrophic Lateral Sclerosis (ALS) is recognized as a very complex disease. As we have learned in the past 20 years from studies in patients and in models based on the expression of mutant SOD1, ALS is not a purely motor neuron disease as previously thought. While undoubtedly motor neurons are lost in patients, a number of alterations in those cell-types that interact functionally with motor neurons (astrocytes, microglia, muscle fibers, oligodendrocytes) take place even long before onset of symptoms. At the same time, disturbance of several, only partly inter-related physiological functions play some role in the onset and progression of the disease. Traditionally, mitochondrial damage and oxidative stress, excitotoxicity, neuroinflammation, altered axonal transport, ER stress, protein aggregation and defective removal of toxic proteins have been considered as key factors in the pathogenesis of ALS, with the relatively recent addition of disturbances in RNA metabolism. This complexity makes the search for an effective treatment extremely difficult and prompts further studies to reveal other possible, previously unappreciated aspects of the pathogenesis of ALS. In this review, we focus on previous knowledge on ALS mechanisms as well as new facets emerging from studies on genetic ALS patients and models that may both provide precious information for a novel therapeutic approach.
Keywords: ALS; RNA metabolism; amyotrophic lateral sclerosis; motor neuron; protein aggregation.
© 2016 International Society of Neuropathology.
Figures
Similar articles
-
Rodent Models of Amyotrophic Lateral Sclerosis.Curr Protoc Pharmacol. 2015 Jun 1;69:5.67.1-5.67.21. doi: 10.1002/0471141755.ph0567s69. Curr Protoc Pharmacol. 2015. PMID: 26344214 Free PMC article. Review.
-
SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS.Adv Biol Regul. 2016 Jan;60:95-104. doi: 10.1016/j.jbior.2015.10.006. Epub 2015 Oct 31. Adv Biol Regul. 2016. PMID: 26563614 Review.
-
System xC- is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice.Brain. 2015 Jan;138(Pt 1):53-68. doi: 10.1093/brain/awu312. Epub 2014 Nov 10. Brain. 2015. PMID: 25384799 Free PMC article.
-
Roots to start research in amyotrophic lateral sclerosis: molecular pathways and novel therapeutics for future.Rev Neurosci. 2015;26(2):161-81. doi: 10.1515/revneuro-2014-0057. Rev Neurosci. 2015. PMID: 25720096 Review.
-
Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS.Prog Neurobiol. 2008 May;85(1):94-134. doi: 10.1016/j.pneurobio.2008.01.001. Epub 2008 Jan 16. Prog Neurobiol. 2008. PMID: 18282652 Review.
Cited by
-
FETR-ALS Study Protocol: A Randomized Clinical Trial of Fecal Microbiota Transplantation in Amyotrophic Lateral Sclerosis.Front Neurol. 2019 Sep 20;10:1021. doi: 10.3389/fneur.2019.01021. eCollection 2019. Front Neurol. 2019. PMID: 31620079 Free PMC article.
-
The Role of Cyclo(His-Pro) in Neurodegeneration.Int J Mol Sci. 2016 Aug 12;17(8):1332. doi: 10.3390/ijms17081332. Int J Mol Sci. 2016. PMID: 27529240 Free PMC article. Review.
-
Functional interaction between FUS and SMN underlies SMA-like splicing changes in wild-type hFUS mice.Sci Rep. 2017 May 17;7(1):2033. doi: 10.1038/s41598-017-02195-0. Sci Rep. 2017. PMID: 28515487 Free PMC article.
-
Combined Treatment with Herbal Medicine and Drug Ameliorates Inflammation and Metabolic Abnormalities in the Liver of an Amyotrophic Lateral Sclerosis Mouse Model.Antioxidants (Basel). 2022 Jan 17;11(1):173. doi: 10.3390/antiox11010173. Antioxidants (Basel). 2022. PMID: 35052677 Free PMC article.
-
Profile of Arachidonic Acid-Derived Inflammatory Markers and Its Modulation by Nitro-Oleic Acid in an Inherited Model of Amyotrophic Lateral Sclerosis.Front Mol Neurosci. 2018 Apr 30;11:131. doi: 10.3389/fnmol.2018.00131. eCollection 2018. Front Mol Neurosci. 2018. PMID: 29760648 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
