

On the reliability of peptide nonplanarity seen in ultra-high resolution crystal structures
- PMID: 26779991
- PMCID: PMC4941219
- DOI: 10.1002/pro.2883
On the reliability of peptide nonplanarity seen in ultra-high resolution crystal structures
Abstract
Ultra-high resolution protein crystal structures have been considered as relatively reliable sources for defining details of protein geometry, such as the extent to which the peptide unit deviates from planarity. Chellapa and Rose (Proteins 2015; 83:1687) recently called this into question, reporting that for a dozen representative protein structures determined at ∼ 1 Å resolution, the diffraction data could be equally well fit with models restrained to have highly planar peptides, i.e. having a standard deviation of the ω torsion angles of only ∼ 1° instead of the typically observed value of ∼ 6°. Here, we document both conceptual and practical shortcomings of that study and show that the more tightly restrained models are demonstrably incorrect and do not fit the diffraction data equally well. We emphasize the importance of inspecting electron density maps when investigating the agreement between a model and its experimental data. Overall, this report reinforces that modern standard refinement protocols have been well-conceived and that ultra-high resolution protein crystal structures, when evaluated carefully and used with an awareness of their levels of coordinate uncertainty, are powerful sources of information for providing reliable information about the details of protein geometry.
Keywords: atomic resolution; model validation; peptide nonplanarity; phenix refinement; protein crystallography; protein geometry.
© 2016 The Protein Society.
Figures
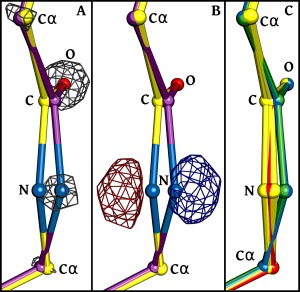
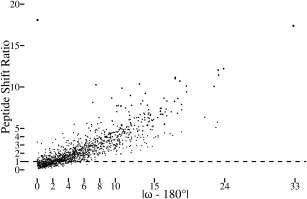
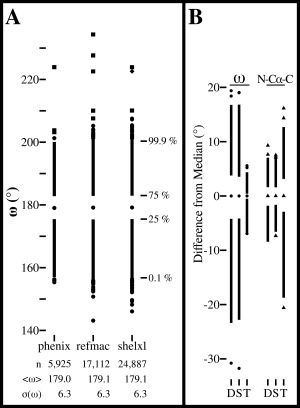
Comment in
-
How planar are planar peptide bonds?Protein Sci. 2016 Apr;25(4):776-7. doi: 10.1002/pro.2901. Epub 2016 Mar 1. Protein Sci. 2016. PMID: 26888078 Free PMC article. No abstract available.
References
-
- Dauter Z, Lamzin VS, Wilson KS (1995) Proteins at atomic resolution. Curr Opin Struct Biol 5:784–790. - PubMed
-
- Dauter Z, Lamzin VS, Wilson KS (1997) The benefits of atomic resolution. Curr Opin Struct Biol 7:681–688. - PubMed
-
- Cruickshank DWJ (1999) Remarks about protein structure precision. Acta Crystallogr D55:583–601. - PubMed
-
- Wilson K, Butterworth S, Dauter Z, Lamzin V, Walsh M, Wodak S, Pontius J, Richelle J, Vaguine A, Sander C, Hooft RWW, Vriend G, Thornton JM, Laskowski RA, MacArthur MW, Dodson EJ, Murshudov G, Oldfield TJ, Kaptein R, Rullmann JAC (1998) Who checks the checkers? Four validation tools applied to eight atomic resolution structures. J Mol Biol 276:417–436. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
