

TGF-β/β2-spectrin/CTCF-regulated tumor suppression in human stem cell disorder Beckwith-Wiedemann syndrome
- PMID: 26784546
- PMCID: PMC4731168
- DOI: 10.1172/JCI80937
TGF-β/β2-spectrin/CTCF-regulated tumor suppression in human stem cell disorder Beckwith-Wiedemann syndrome
Abstract
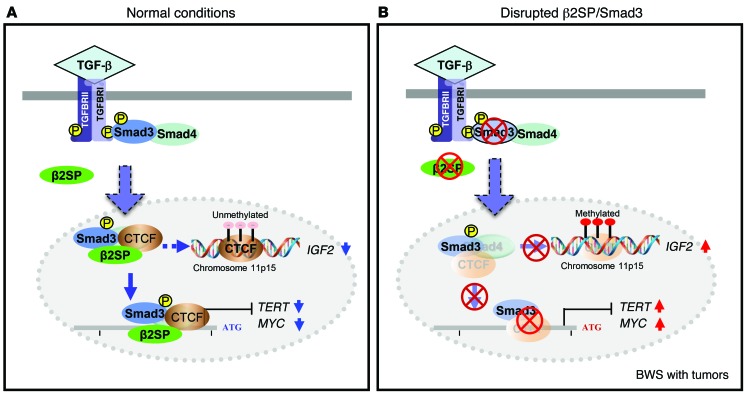
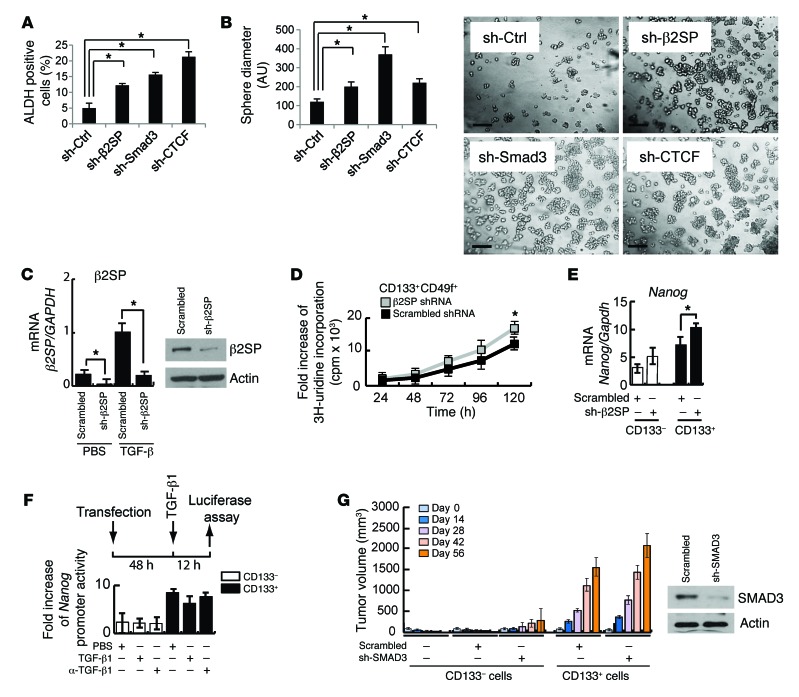
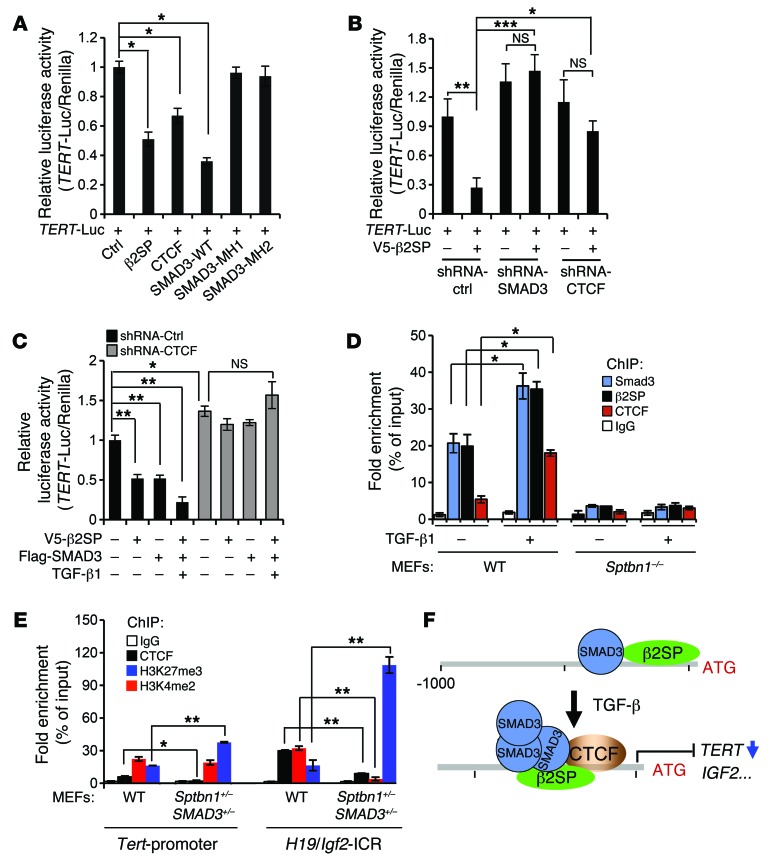
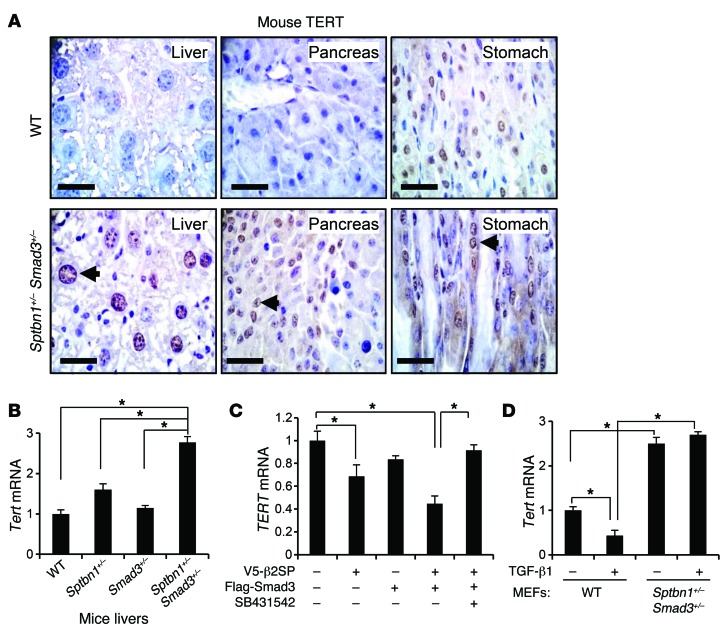
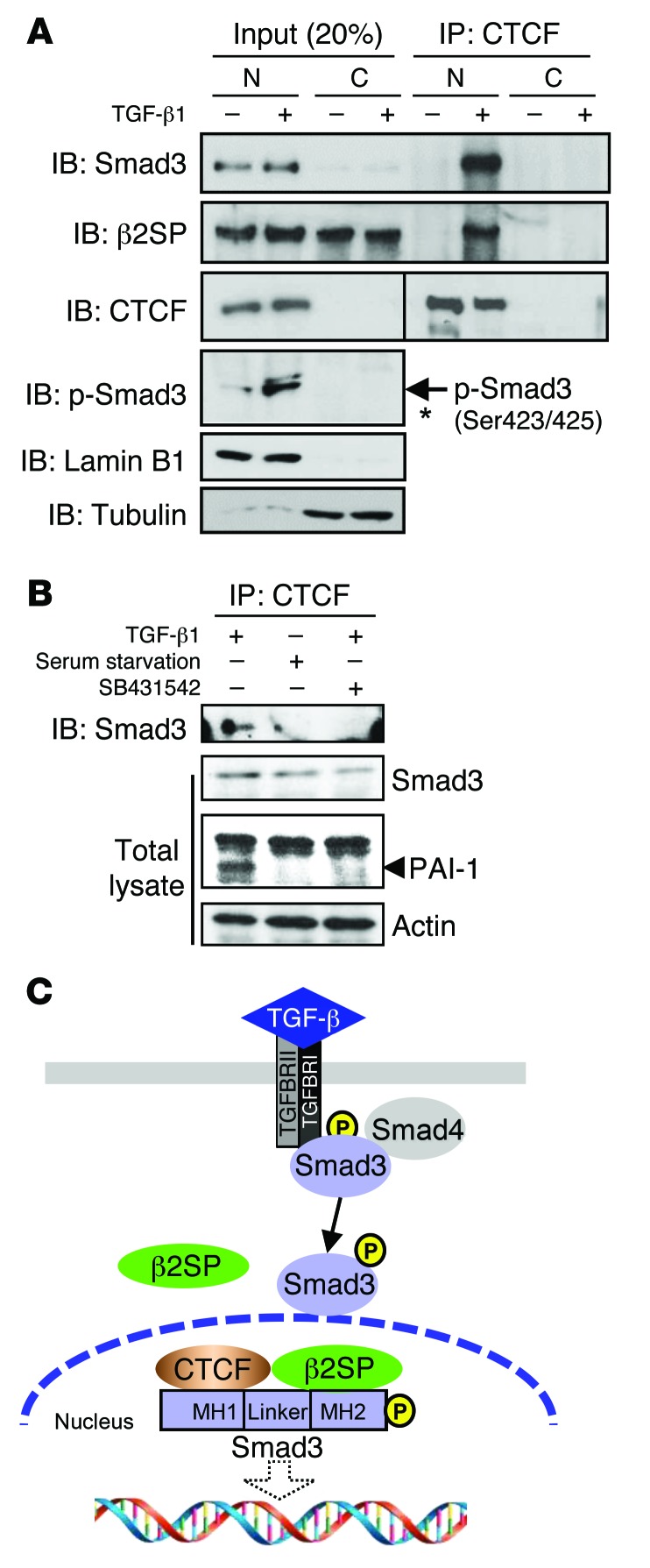
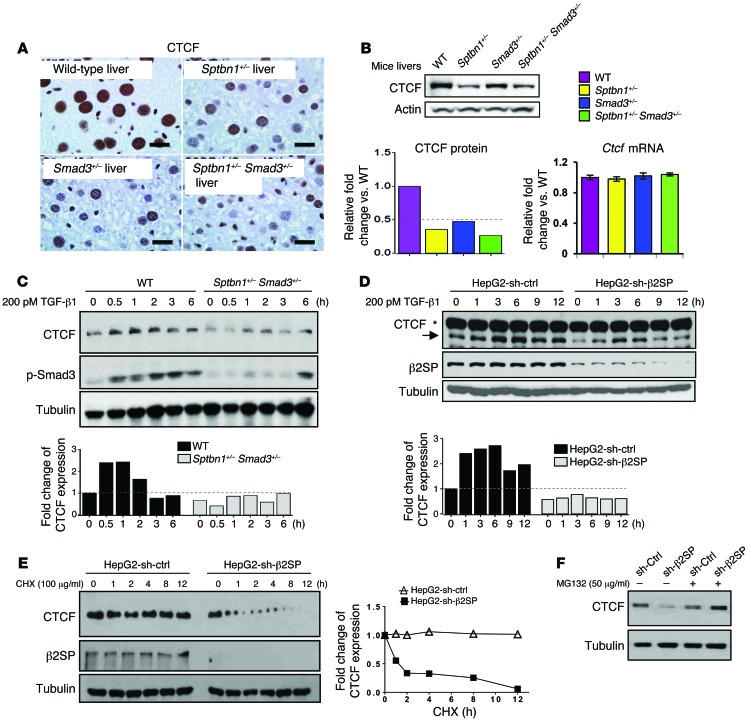
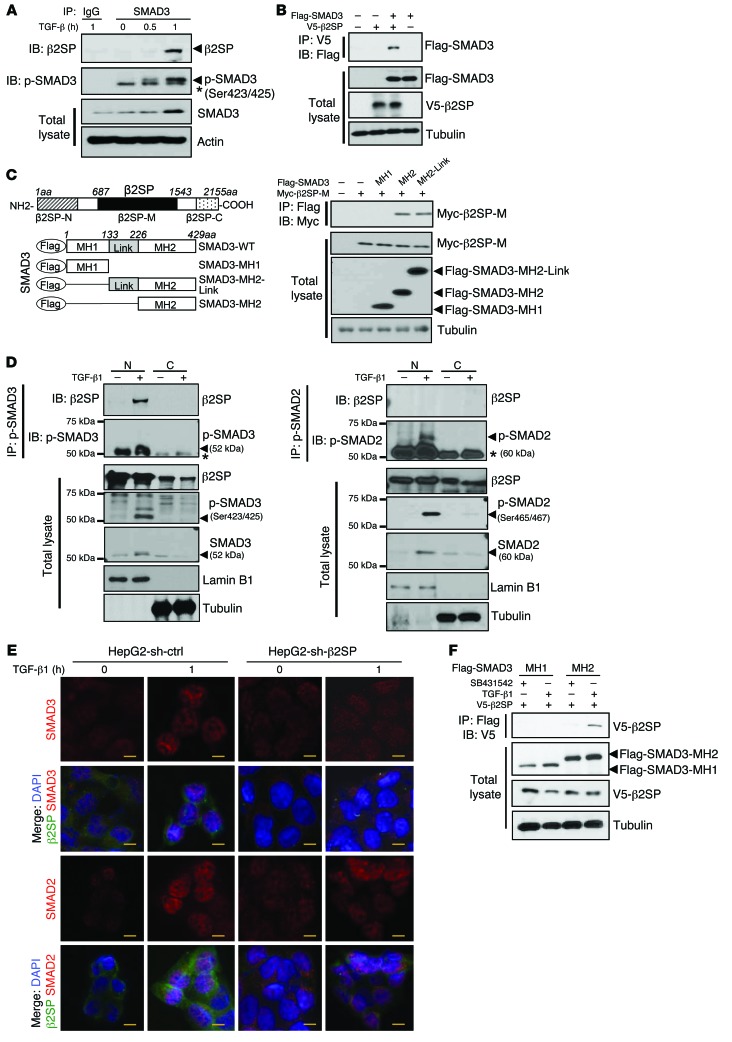
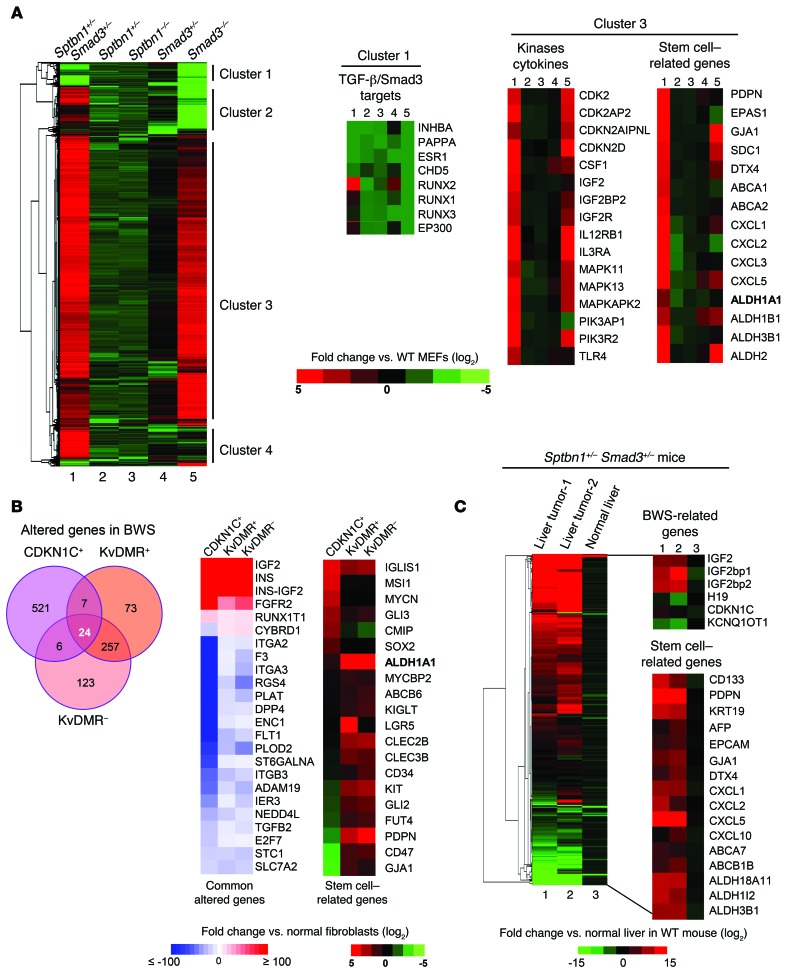
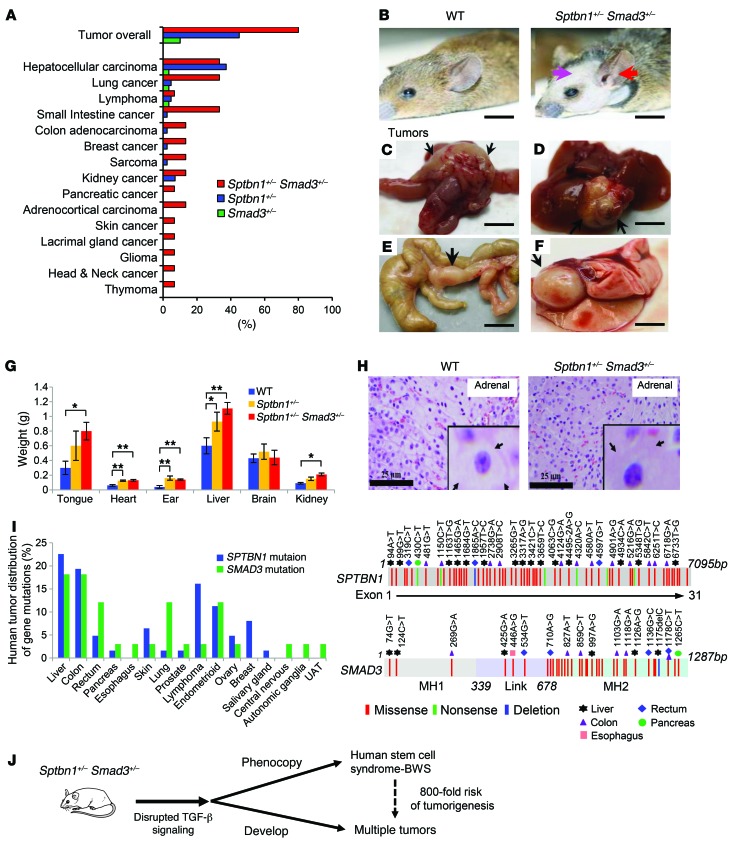
Beckwith-Wiedemann syndrome (BWS) is a human stem cell disorder, and individuals with this disease have a substantially increased risk (~800-fold) of developing tumors. Epigenetic silencing of β2-spectrin (β2SP, encoded by SPTBN1), a SMAD adaptor for TGF-β signaling, is causally associated with BWS; however, a role of TGF-β deficiency in BWS-associated neoplastic transformation is unexplored. Here, we have reported that double-heterozygous Sptbn1+/- Smad3+/- mice, which have defective TGF-β signaling, develop multiple tumors that are phenotypically similar to those of BWS patients. Moreover, tumorigenesis-associated genes IGF2 and telomerase reverse transcriptase (TERT) were overexpressed in fibroblasts from BWS patients and TGF-β-defective mice. We further determined that chromatin insulator CCCTC-binding factor (CTCF) is TGF-β inducible and facilitates TGF-β-mediated repression of TERT transcription via interactions with β2SP and SMAD3. This regulation was abrogated in TGF-β-defective mice and BWS, resulting in TERT overexpression. Imprinting of the IGF2/H19 locus and the CDKN1C/KCNQ1 locus on chromosome 11p15.5 is mediated by CTCF, and this regulation is lost in BWS, leading to aberrant overexpression of growth-promoting genes. Therefore, we propose that loss of CTCF-dependent imprinting of tumor-promoting genes, such as IGF2 and TERT, results from a defective TGF-β pathway and is responsible at least in part for BWS-associated tumorigenesis as well as sporadic human cancers that are frequently associated with SPTBN1 and SMAD3 mutations.
Figures
References
-
- DeBaun MR, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr. 1998;132(3 pt 1):398–400. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- P01 CA130821/CA/NCI NIH HHS/United States
- R01 AA023146/AA/NIAAA NIH HHS/United States
- R01AA023146/AA/NIAAA NIH HHS/United States
- U01 AA018663/AA/NIAAA NIH HHS/United States
- P30 DK56338/DK/NIDDK NIH HHS/United States
- I01 BX001991/BX/BLRD VA/United States
- RC2 AA019392/AA/NIAAA NIH HHS/United States
- R01 HL085218/HL/NHLBI NIH HHS/United States
- R01 CA106614/CA/NCI NIH HHS/United States
- P30 DK056338/DK/NIDDK NIH HHS/United States
- R01 AA018857/AA/NIAAA NIH HHS/United States
- R01CA042857/CA/NCI NIH HHS/United States
- R01AA018857/AA/NIAAA NIH HHS/United States
- P30 DK048522/DK/NIDDK NIH HHS/United States
- R01 CA042857/CA/NCI NIH HHS/United States
- P50 AA011999/AA/NIAAA NIH HHS/United States
- P50AA011999/AA/NIAAA NIH HHS/United States
- U01AA018663/AA/NIAAA NIH HHS/United States
- R01CA106614/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
