

Improved proteostasis in the secretory pathway rescues Alzheimer's disease in the mouse
- PMID: 26787453
- PMCID: PMC4805081
- DOI: 10.1093/brain/awv385
Improved proteostasis in the secretory pathway rescues Alzheimer's disease in the mouse
Abstract
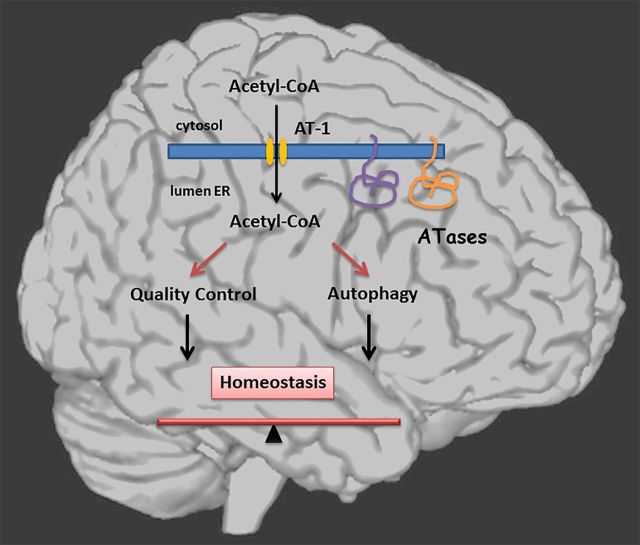
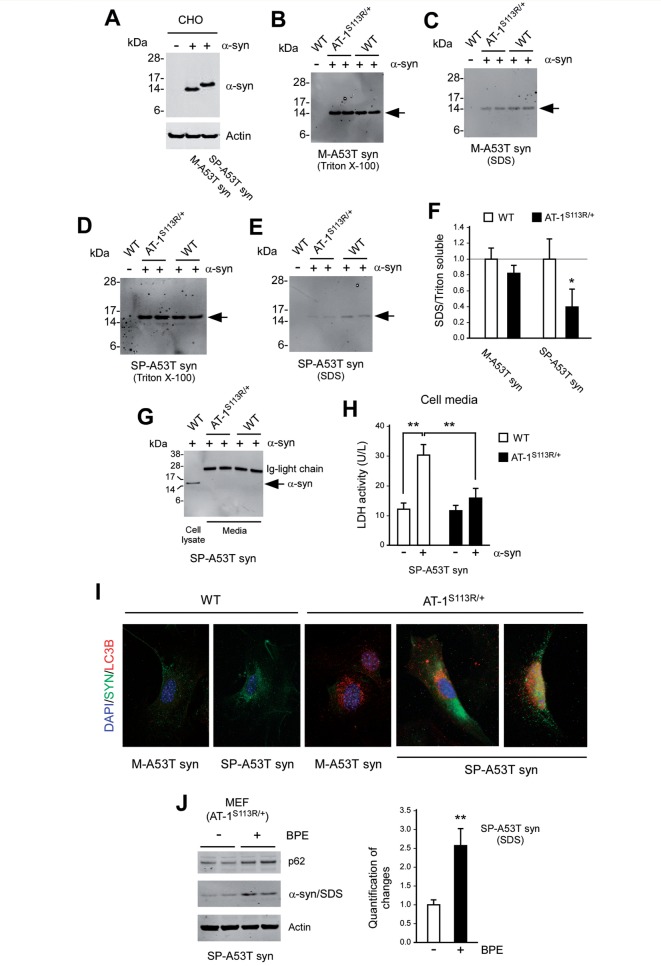
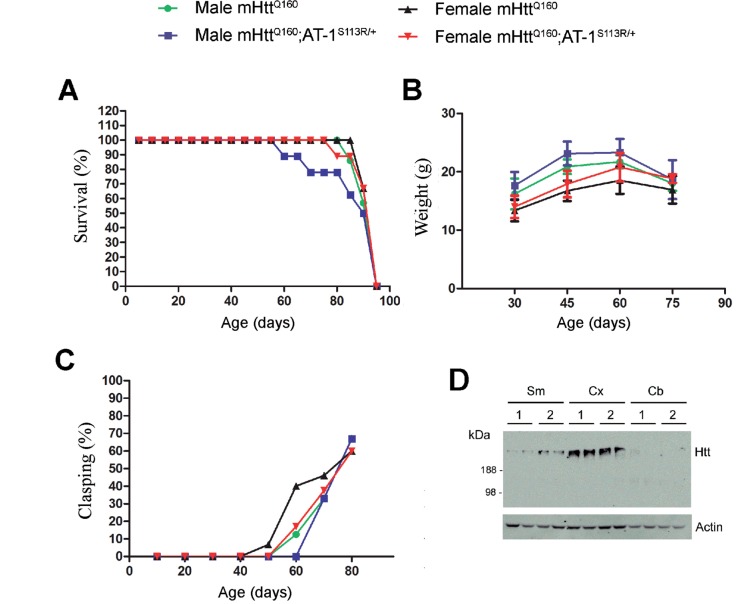
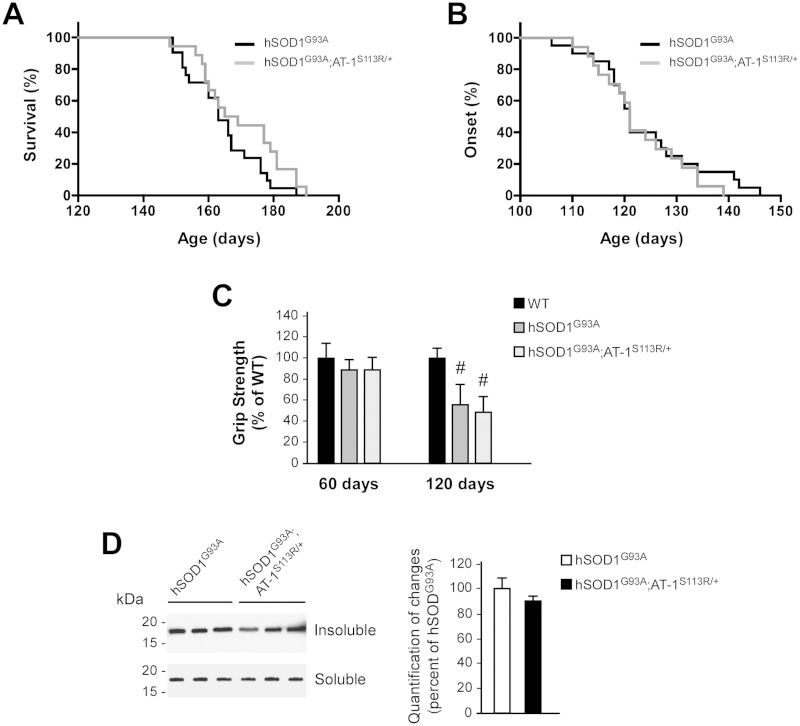
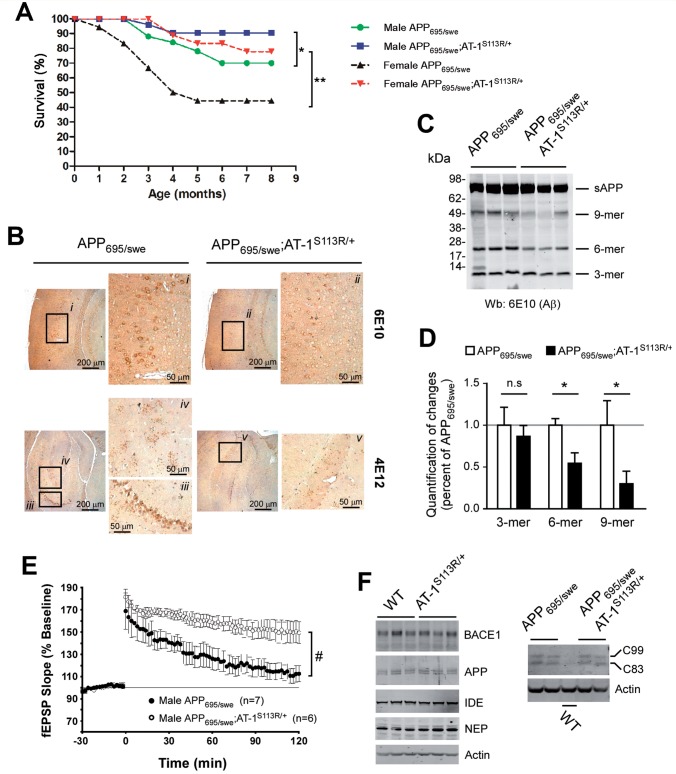
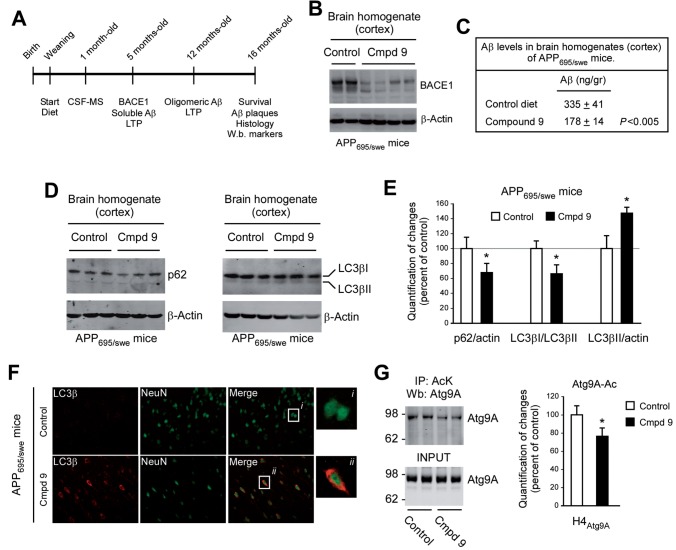
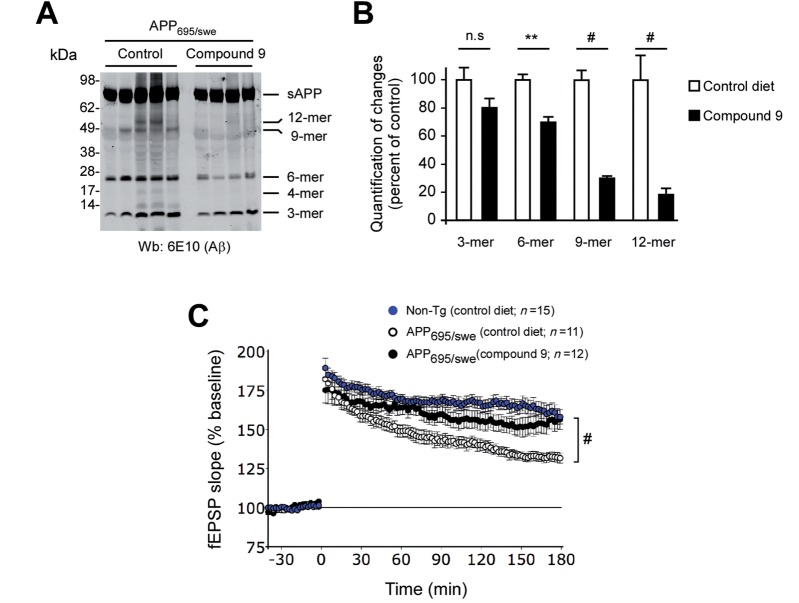
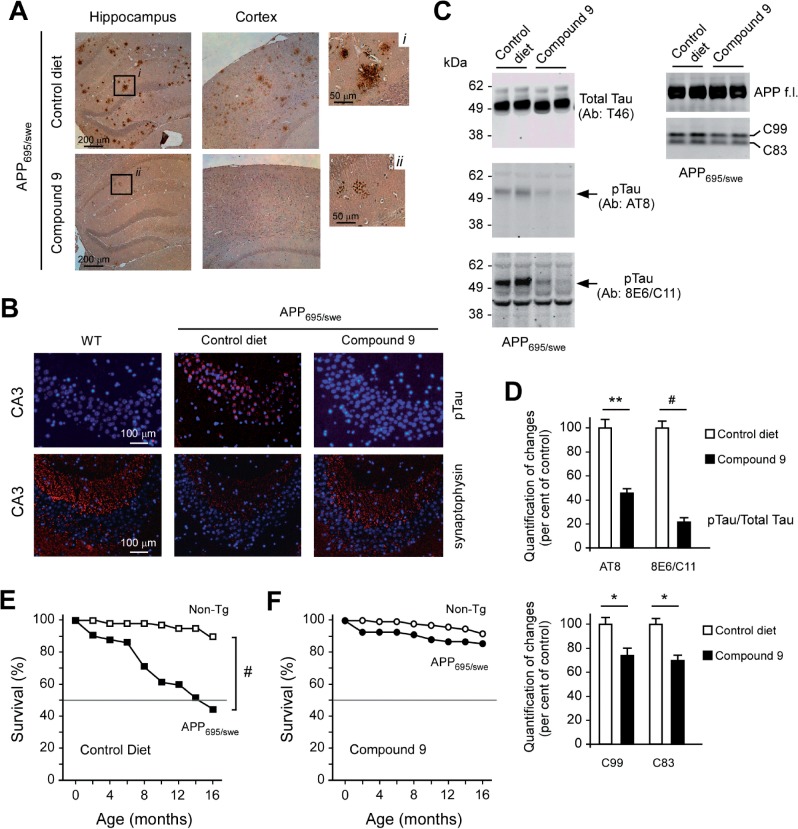
The aberrant accumulation of toxic protein aggregates is a key feature of many neurodegenerative diseases, including Huntington's disease, amyotrophic lateral sclerosis and Alzheimer's disease. As such, improving normal proteostatic mechanisms is an active target for biomedical research. Although they share common pathological features, protein aggregates form in different subcellular locations. Nε-lysine acetylation in the lumen of the endoplasmic reticulum has recently emerged as a new mechanism to regulate the induction of autophagy. The endoplasmic reticulum acetylation machinery includes AT-1/SLC33A1, a membrane transporter that translocates acetyl-CoA from the cytosol into the endoplasmic reticulum lumen, and ATase1 and ATase2, two acetyltransferases that acetylate endoplasmic reticulum cargo proteins. Here, we used a mutant form of α-synuclein to show that inhibition of the endoplasmic reticulum acetylation machinery specifically improves autophagy-mediated disposal of toxic protein aggregates that form within the secretory pathway, but not those that form in the cytosol. Consequently, haploinsufficiency of AT-1/SLC33A1 in the mouse rescued Alzheimer's disease, but not Huntington's disease or amyotrophic lateral sclerosis. In fact, intracellular toxic protein aggregates in Alzheimer's disease form within the secretory pathway while in Huntington's disease and amyotrophic lateral sclerosis they form in different cellular compartments. Furthermore, biochemical inhibition of ATase1 and ATase2 was also able to rescue the Alzheimer's disease phenotype in a mouse model of the disease. Specifically, we observed reduced levels of soluble amyloid-β aggregates, reduced amyloid-β pathology, reduced phosphorylation of tau, improved synaptic plasticity, and increased lifespan of the animals. In conclusion, our results indicate that Nε-lysine acetylation in the endoplasmic reticulum lumen regulates normal proteostasis of the secretory pathway; they also support therapies targeting endoplasmic reticulum acetyltransferases, ATase1 and ATase2, for a subset of chronic degenerative diseases.
Keywords: AT-1/SLC33A1; ATase; Alzheimer’s disease; autophagy; lysine acetylation; proteostasis.
© The Author (2016). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Comment in
-
Targeting endoplasmic reticulum acetylation to restore proteostasis in Alzheimer's disease.Brain. 2016 Mar;139(Pt 3):650-2. doi: 10.1093/brain/awv401. Brain. 2016. PMID: 26917585 No abstract available.
References
-
- Bhattacharya A, Bokov A, Muller FL, Jernigan AL, Maslin K, Diaz V, et al. . Dietary restriction but not rapamycin extends disease onset and survival of the H46R/H48Q mouse model of ALS . Neurobiol Aging 2012. ; 33 : 1829 – 32 . - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
