

Rescue of Impaired mGluR5-Driven Endocannabinoid Signaling Restores Prefrontal Cortical Output to Inhibit Pain in Arthritic Rats
- PMID: 26791214
- PMCID: PMC4719019
- DOI: 10.1523/JNEUROSCI.4047-15.2016
Rescue of Impaired mGluR5-Driven Endocannabinoid Signaling Restores Prefrontal Cortical Output to Inhibit Pain in Arthritic Rats
Abstract
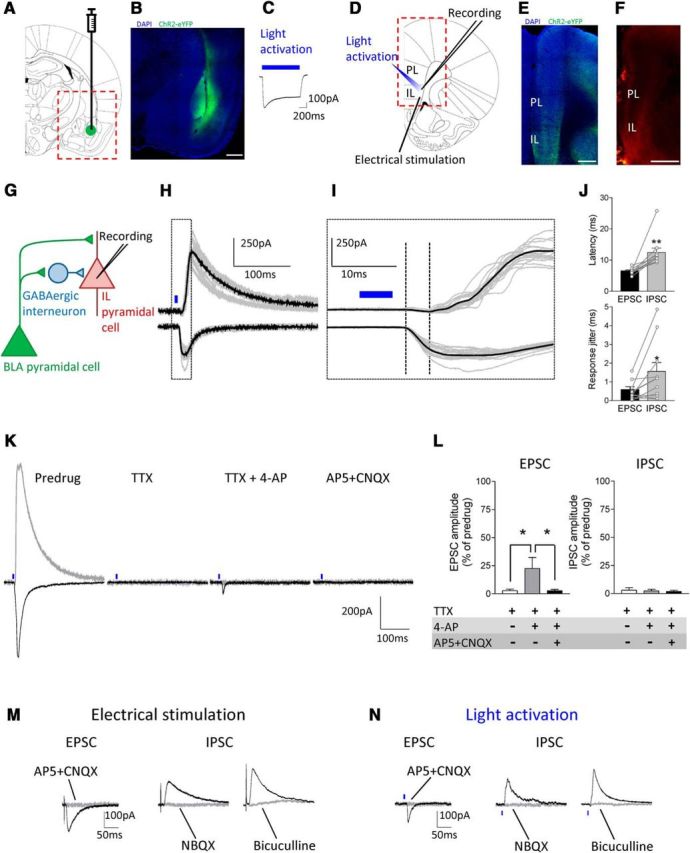
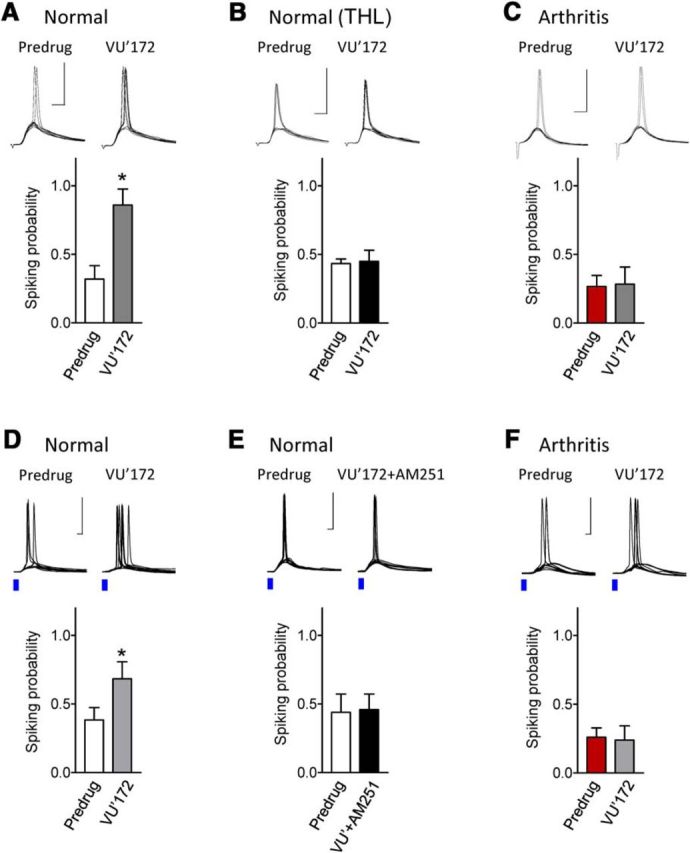
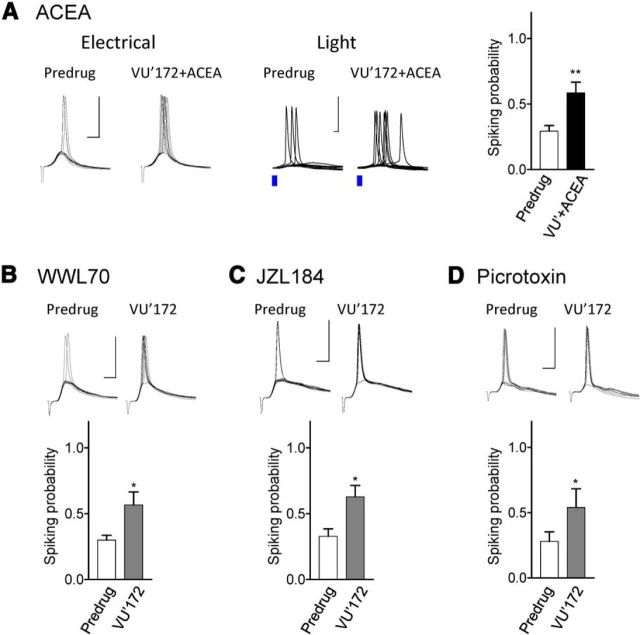
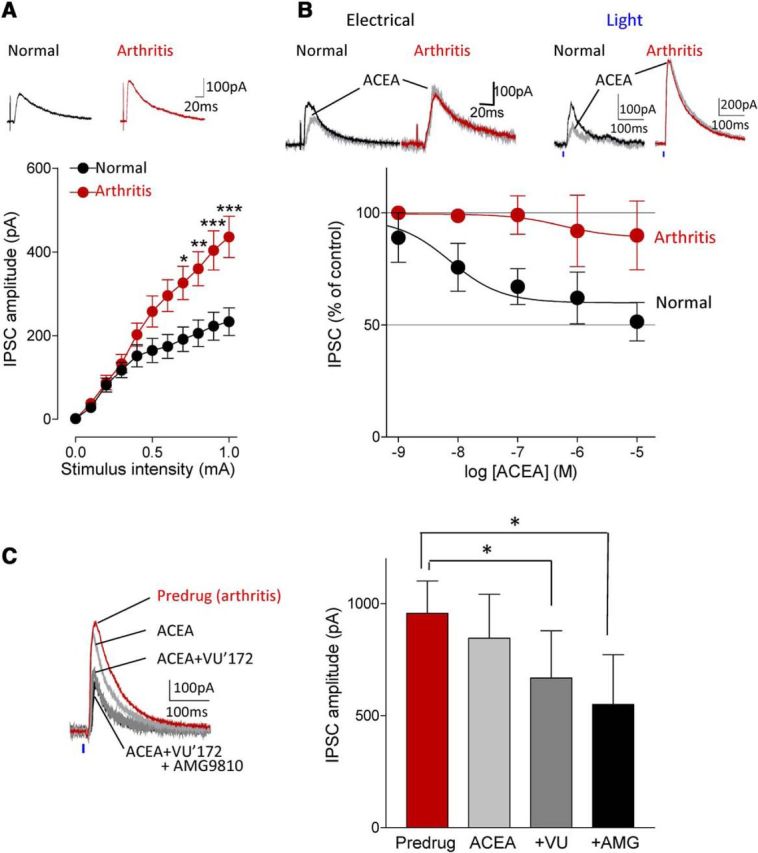
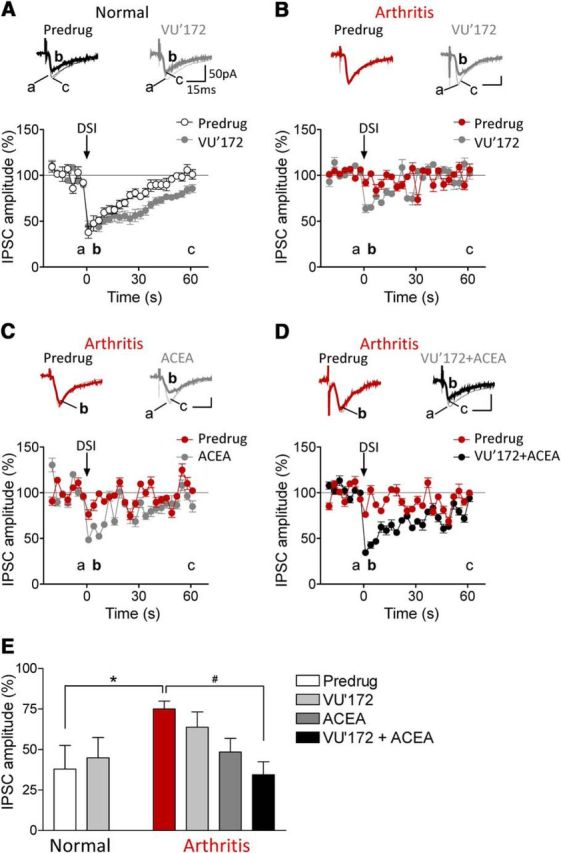
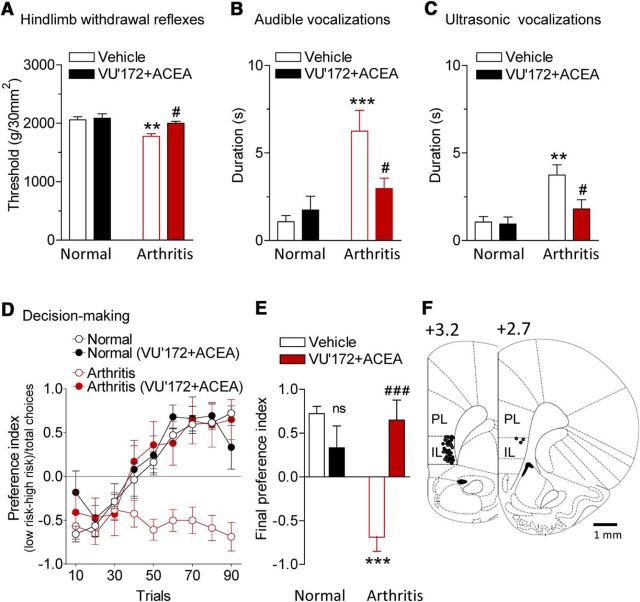
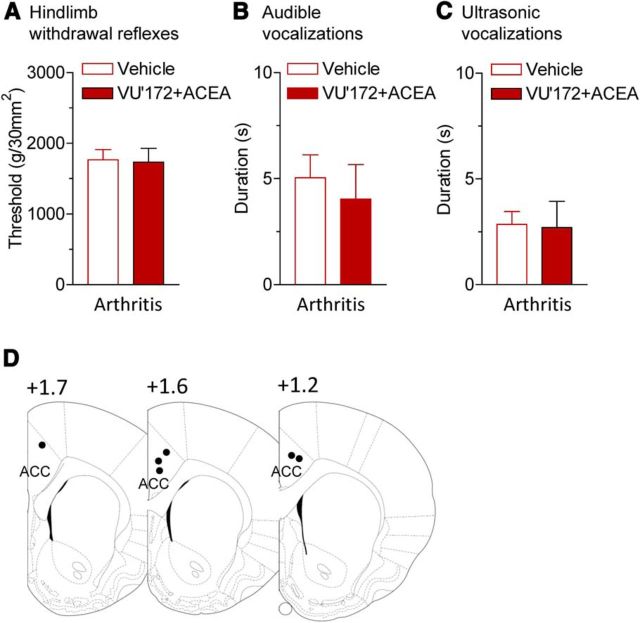
The medial prefrontal cortex (mPFC) serves executive functions that are impaired in neuropsychiatric disorders and pain. Underlying mechanisms remain to be determined. Here we advance the novel concept that metabotropic glutamate receptor 5 (mGluR5) fails to engage endocannabinoid (2-AG) signaling to overcome abnormal synaptic inhibition in pain, but restoring endocannabinoid signaling allows mGluR5 to increase mPFC output hence inhibit pain behaviors and mitigate cognitive deficits. Whole-cell patch-clamp recordings were made from layer V pyramidal cells in the infralimbic mPFC in rat brain slices. Electrical and optogenetic stimulations were used to analyze amygdala-driven mPFC activity. A selective mGluR5 activator (VU0360172) increased pyramidal output through an endocannabinoid-dependent mechanism because intracellular inhibition of the major 2-AG synthesizing enzyme diacylglycerol lipase or blockade of CB1 receptors abolished the facilitatory effect of VU0360172. In an arthritis pain model mGluR5 activation failed to overcome abnormal synaptic inhibition and increase pyramidal output. mGluR5 function was rescued by restoring 2-AG-CB1 signaling with a CB1 agonist (ACEA) or inhibitors of postsynaptic 2-AG hydrolyzing enzyme ABHD6 (intracellular WWL70) and monoacylglycerol lipase MGL (JZL184) or by blocking GABAergic inhibition with intracellular picrotoxin. CB1-mediated depolarization-induced suppression of synaptic inhibition (DSI) was also impaired in the pain model but could be restored by coapplication of VU0360172 and ACEA. Stereotaxic coadministration of VU0360172 and ACEA into the infralimbic, but not anterior cingulate, cortex mitigated decision-making deficits and pain behaviors of arthritic animals. The results suggest that rescue of impaired endocannabinoid-dependent mGluR5 function in the mPFC can restore mPFC output and cognitive functions and inhibit pain. Significance statement: Dysfunctions in prefrontal cortical interactions with subcortical brain regions, such as the amygdala, are emerging as important players in neuropsychiatric disorders and pain. This study identifies a novel mechanism and rescue strategy for impaired medial prefrontal cortical function in an animal model of arthritis pain. Specifically, an integrative approach of optogenetics, pharmacology, electrophysiology, and behavior is used to advance the novel concept that a breakdown of metabotropic glutamate receptor subtype mGluR5 and endocannabinoid signaling in infralimbic pyramidal cells fails to control abnormal amygdala-driven synaptic inhibition in the arthritis pain model. Restoring endocannabinoid signaling allows mGluR5 activation to increase infralimbic output hence inhibit pain behaviors and mitigate pain-related cognitive deficits.
Keywords: amygdala; cannabinoids; mGluR; pain; plasticity; prefrontal cortex.
Copyright © 2016 the authors 0270-6474/16/360837-14$15.00/0.
Figures
References
-
- Bisogno T, Cascio MG, Saha B, Mahadevan A, Urbani P, Minassi A, Appendino G, Saturnino C, Martin B, Razdan R, Di Marzo V. Development of the first potent and specific inhibitors of endocannabinoid biosynthesis. Biochim Biophys Acta. 2006;1761:205–212. doi: 10.1016/j.bbalip.2005.12.009. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical