

Quantifying prion disease penetrance using large population control cohorts
- PMID: 26791950
- PMCID: PMC4774245
- DOI: 10.1126/scitranslmed.aad5169
Quantifying prion disease penetrance using large population control cohorts
Abstract
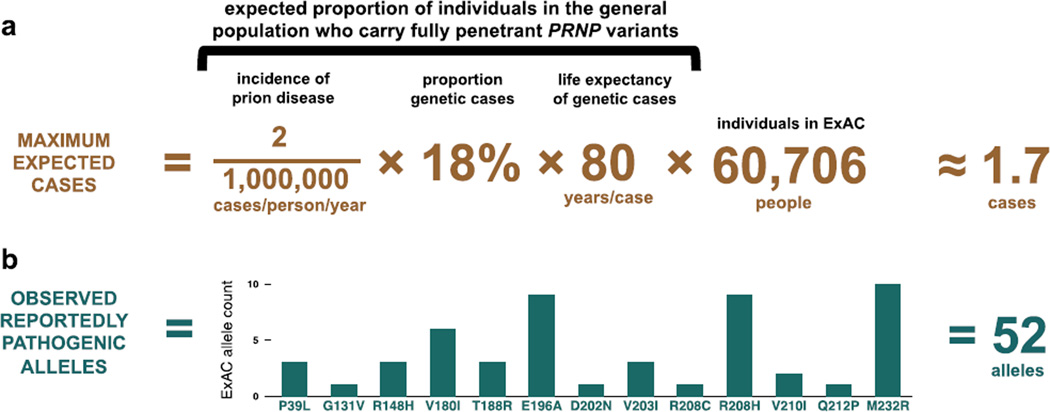
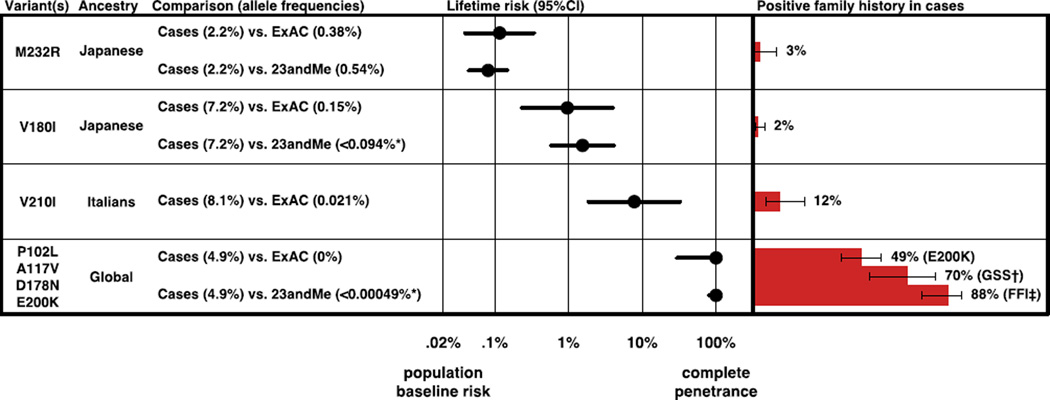
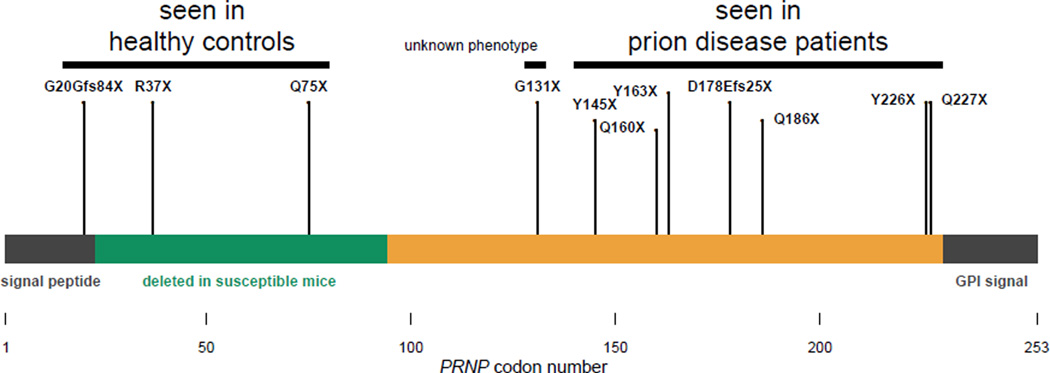
More than 100,000 genetic variants are reported to cause Mendelian disease in humans, but the penetrance-the probability that a carrier of the purported disease-causing genotype will indeed develop the disease-is generally unknown. We assess the impact of variants in the prion protein gene (PRNP) on the risk of prion disease by analyzing 16,025 prion disease cases, 60,706 population control exomes, and 531,575 individuals genotyped by 23andMe Inc. We show that missense variants in PRNP previously reported to be pathogenic are at least 30 times more common in the population than expected on the basis of genetic prion disease prevalence. Although some of this excess can be attributed to benign variants falsely assigned as pathogenic, other variants have genuine effects on disease susceptibility but confer lifetime risks ranging from <0.1 to ~100%. We also show that truncating variants in PRNP have position-dependent effects, with true loss-of-function alleles found in healthy older individuals, a finding that supports the safety of therapeutic suppression of prion protein expression.
Copyright © 2016, American Association for the Advancement of Science.
Figures

Comment in
-
"Big Data" Gets Personal.Sci Transl Med. 2016 Jan 20;8(322):322fs3-3fs3. doi: 10.1126/scitranslmed.aad9460. Sci Transl Med. 2016. PMID: 26791946
-
Disease genetics: Prion variant pathogenicity through large-scale population sequencing.Nat Rev Genet. 2016 Mar;17(3):127. doi: 10.1038/nrg.2016.9. Epub 2016 Feb 1. Nat Rev Genet. 2016. PMID: 26831349 No abstract available.
References
-
- Amberger J, Bocchini C, Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®) Hum. Mutat. 2011;32:564–567. - PubMed
-
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Harrell TM, McMillin MJ, Wiszniewski W, Gambin T, Coban Akdemir ZH, Doheny K, Scott AF, Avramopoulos D, Chakravarti A, Hoover-Fong J, Mathews D, Witmer PD, Ling H, Hetrick K, Watkins L, Patterson KE, Reinier F, Blue E, Muzny D, Kircher M, Bilguvar K, López-Giráldez F, Sutton VR, Tabor HK, Leal SM, Gunel M, Mane S, Gibbs RA, Boerwinkle E, Hamosh A, Shendure J, Lupski JR, Lifton RP, Valle D, Nickerson DA, Centers for Mendelian Genomics. Bamshad MJ. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 2015;97:199–215. - PMC - PubMed
-
- Begg CB. On the use of familial aggregation in population-based case probands for calculating penetrance. J. Natl. Cancer Inst. 2002;94:1221–1226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DK093757/DK/NIDDK NIH HHS/United States
- R01GM104371/GM/NIGMS NIH HHS/United States
- R01 MH095034/MH/NIMH NIH HHS/United States
- R01 DK072193/DK/NIDDK NIH HHS/United States
- Department of Health/United Kingdom
- UR8/CCU515004/PHS HHS/United States
- U01 DK062370/DK/NIDDK NIH HHS/United States
- U54 DK105566/DK/NIDDK NIH HHS/United States
- U54DK105566/DK/NIDDK NIH HHS/United States
- P30 DK020572/DK/NIDDK NIH HHS/United States
- UR8 CI515004/CI/NCPDCID CDC HHS/United States
- R01 GM104371/GM/NIGMS NIH HHS/United States
- F32 GM115208/GM/NIGMS NIH HHS/United States
