

Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin
- PMID: 26792326
- PMCID: PMC6514469
- DOI: 10.1242/jeb.125930
Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin
Abstract
Hypertrophic cardiomyopathy is the most frequently occurring inherited cardiovascular disease, with a prevalence of more than one in 500 individuals worldwide. Genetically acquired dilated cardiomyopathy is a related disease that is less prevalent. Both are caused by mutations in the genes encoding the fundamental force-generating protein machinery of the cardiac muscle sarcomere, including human β-cardiac myosin, the motor protein that powers ventricular contraction. Despite numerous studies, most performed with non-human or non-cardiac myosin, there is no clear consensus about the mechanism of action of these mutations on the function of human β-cardiac myosin. We are using a recombinantly expressed human β-cardiac myosin motor domain along with conventional and new methodologies to characterize the forces and velocities of the mutant myosins compared with wild type. Our studies are extending beyond myosin interactions with pure actin filaments to include the interaction of myosin with regulated actin filaments containing tropomyosin and troponin, the roles of regulatory light chain phosphorylation on the functions of the system, and the possible roles of myosin binding protein-C and titin, important regulatory components of both cardiac and skeletal muscles.
Keywords: Cardiac myosin; Cardiomyopathy mutations; Force–velocity curves.
© 2016. Published by The Company of Biologists Ltd.
Conflict of interest statement
J.A.S. is a founder of and owns shares in Cytokinetics, Inc., and MyoKardia, Inc., biotech companies that are developing therapeutics that target the sarcomere.
Figures
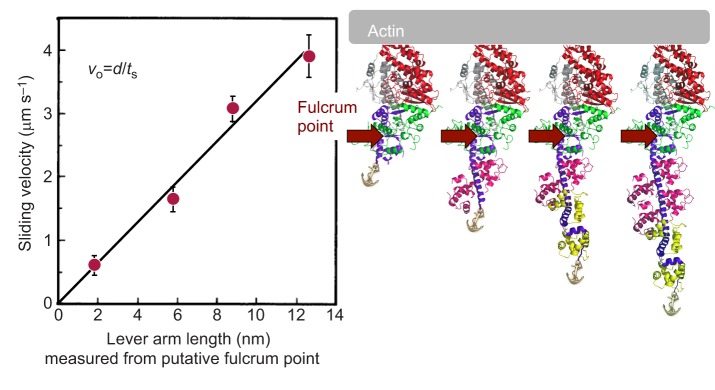
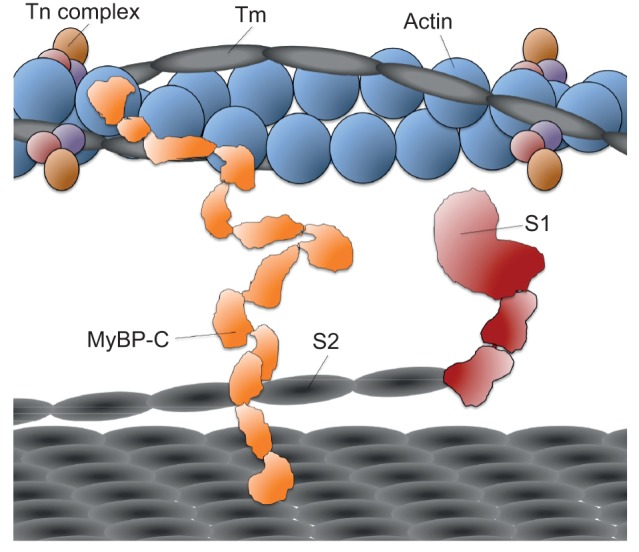
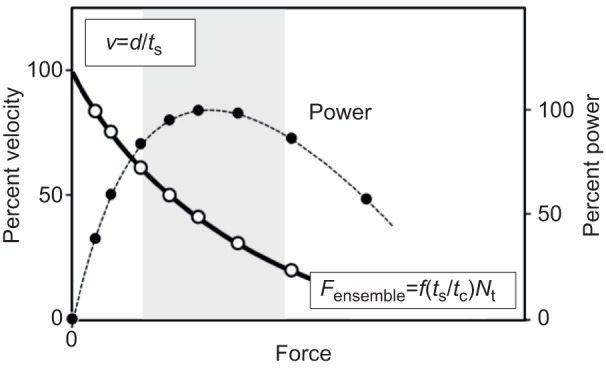
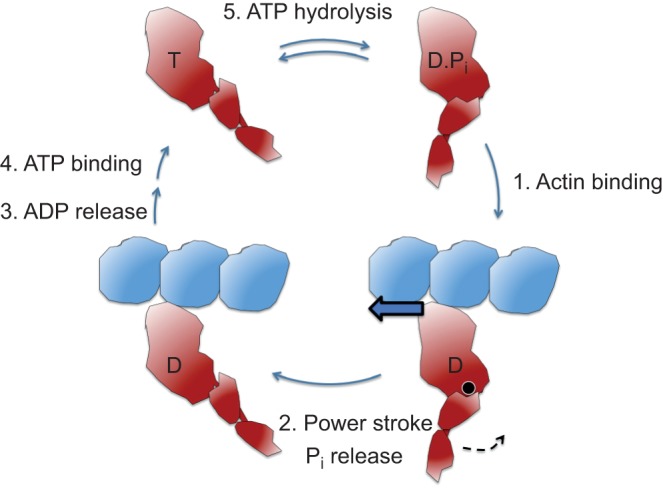
Similar articles
-
Hypertrophic cardiomyopathy β-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state.Proc Natl Acad Sci U S A. 2021 Jun 15;118(24):e2025030118. doi: 10.1073/pnas.2025030118. Proc Natl Acad Sci U S A. 2021. PMID: 34117120 Free PMC article.
-
Early-Onset Hypertrophic Cardiomyopathy Mutations Significantly Increase the Velocity, Force, and Actin-Activated ATPase Activity of Human β-Cardiac Myosin.Cell Rep. 2016 Dec 13;17(11):2857-2864. doi: 10.1016/j.celrep.2016.11.040. Cell Rep. 2016. PMID: 27974200 Free PMC article.
-
Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human β-cardiac myosin.PLoS One. 2013 Dec 18;8(12):e83403. doi: 10.1371/journal.pone.0083403. eCollection 2013. PLoS One. 2013. PMID: 24367593 Free PMC article.
-
[Familial hypertrophic cardiomyopathy: genes, mutations and animal models. A review].Invest Clin. 2004 Mar;45(1):69-99. Invest Clin. 2004. PMID: 15058760 Review. Spanish.
-
Molecular basis of hypertrophic and dilated cardiomyopathy.Tex Heart Inst J. 1994;21(1):6-15. Tex Heart Inst J. 1994. PMID: 8180512 Free PMC article. Review.
Cited by
-
X-ray Crystallographic and Molecular Dynamic Analyses of Drosophila melanogaster Embryonic Muscle Myosin Define Domains Responsible for Isoform-Specific Properties.J Mol Biol. 2020 Jan 17;432(2):427-447. doi: 10.1016/j.jmb.2019.11.013. Epub 2019 Nov 29. J Mol Biol. 2020. PMID: 31786266 Free PMC article.
-
Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations.Pflugers Arch. 2019 May;471(5):701-717. doi: 10.1007/s00424-019-02259-2. Epub 2019 Feb 15. Pflugers Arch. 2019. PMID: 30767072 Free PMC article. Review.
-
Hereditary heart disease: pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains.Pflugers Arch. 2019 May;471(5):683-699. doi: 10.1007/s00424-019-02257-4. Epub 2019 Jan 31. Pflugers Arch. 2019. PMID: 30706179 Free PMC article. Review.
-
β-Cardiac myosin hypertrophic cardiomyopathy mutations release sequestered heads and increase enzymatic activity.Nat Commun. 2019 Jun 18;10(1):2685. doi: 10.1038/s41467-019-10555-9. Nat Commun. 2019. PMID: 31213605 Free PMC article.
-
Modulating Beta-Cardiac Myosin Function at the Molecular and Tissue Levels.Front Physiol. 2017 Jan 9;7:659. doi: 10.3389/fphys.2016.00659. eCollection 2016. Front Physiol. 2017. PMID: 28119616 Free PMC article.
References
-
- Alfares A. A., Kelly M. A., McDermott G., Funke B. H., Lebo M. S., Baxter S. B., Shen J., McLaughlin H. M., Clark E. H., Babb L. J. et al. (2015). Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet. Med 17, 880-888. 10.1038/gim.2014.205 - DOI - PubMed
-
- Bing W., Knott A. and Marston S. B. (2000). A simple method for measuring the relative force exerted by myosin on actin filaments in the in vitro motility assay: evidence that tropomyosin and troponin increase force in single thin filaments. Biochem. J. 350, 693-699. 10.1042/bj3500693 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
