Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation
- PMID: 26795250
- PMCID: PMC5330663
- DOI: 10.1016/j.immuni.2015.12.006
Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation
Abstract
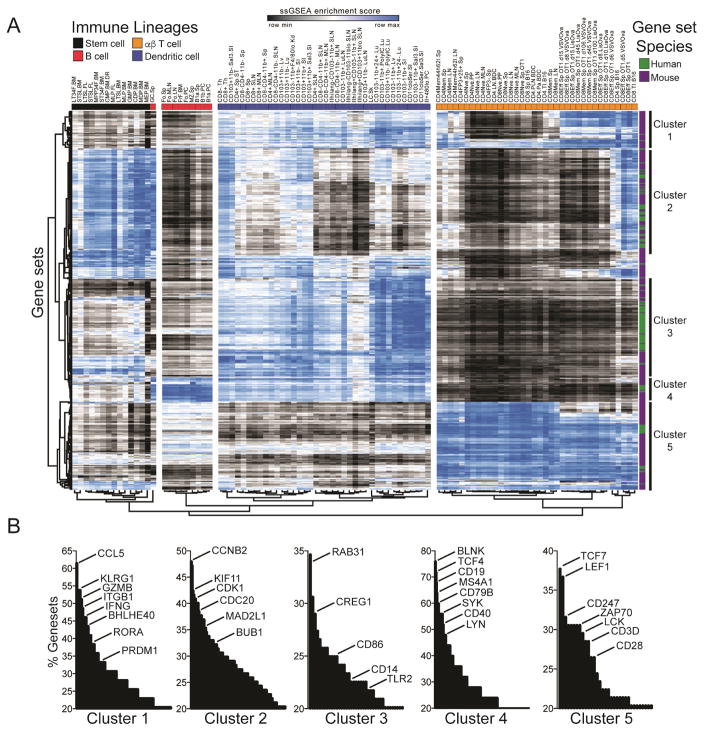
Gene-expression profiling has become a mainstay in immunology, but subtle changes in gene networks related to biological processes are hard to discern when comparing various datasets. For instance, conservation of the transcriptional response to sepsis in mouse models and human disease remains controversial. To improve transcriptional analysis in immunology, we created ImmuneSigDB: a manually annotated compendium of ∼5,000 gene-sets from diverse cell states, experimental manipulations, and genetic perturbations in immunology. Analysis using ImmuneSigDB identified signatures induced in activated myeloid cells and differentiating lymphocytes that were highly conserved between humans and mice. Sepsis triggered conserved patterns of gene expression in humans and mouse models. However, we also identified species-specific biological processes in the sepsis transcriptional response: although both species upregulated phagocytosis-related genes, a mitosis signature was specific to humans. ImmuneSigDB enables granular analysis of transcriptomic data to improve biological understanding of immune processes of the human and mouse immune systems.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures