

In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity
- PMID: 26797105
- PMCID: PMC4726175
- DOI: 10.1038/srep19848
In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity
Abstract
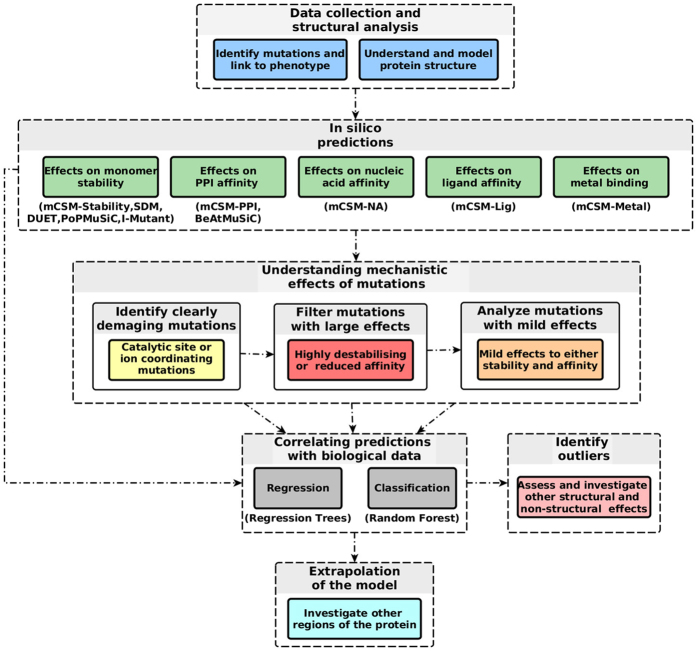
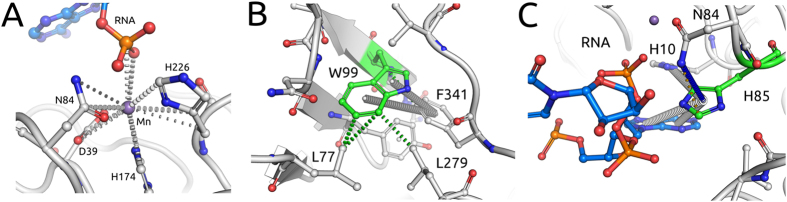
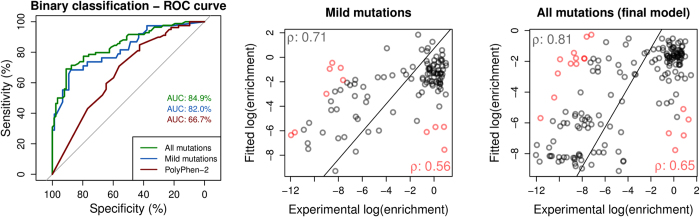
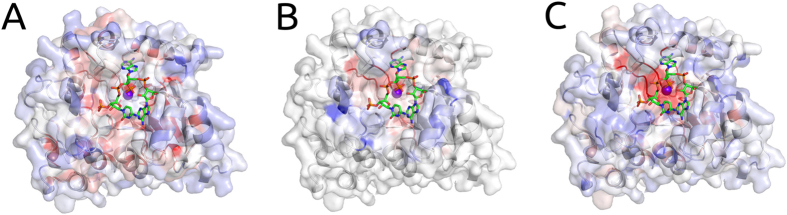
Despite interest in associating polymorphisms with clinical or experimental phenotypes, functional interpretation of mutation data has lagged behind generation of data from modern high-throughput techniques and the accurate prediction of the molecular impact of a mutation remains a non-trivial task. We present here an integrated knowledge-driven computational workflow designed to evaluate the effects of experimental and disease missense mutations on protein structure and interactions. We exemplify its application with analyses of saturation mutagenesis of DBR1 and Gal4 and show that the experimental phenotypes for over 80% of the mutations correlate well with predicted effects of mutations on protein stability and RNA binding affinity. We also show that analysis of mutations in VHL using our workflow provides valuable insights into the effects of mutations, and their links to the risk of developing renal carcinoma. Taken together the analyses of the three examples demonstrate that structural bioinformatics tools, when applied in a systematic, integrated way, can rapidly analyse a given system to provide a powerful approach for predicting structural and functional effects of thousands of mutations in order to reveal molecular mechanisms leading to a phenotype. Missense or non-synonymous mutations are nucleotide substitutions that alter the amino acid sequence of a protein. Their effects can range from modifying transcription, translation, processing and splicing, localization, changing stability of the protein, altering its dynamics or interactions with other proteins, nucleic acids and ligands, including small molecules and metal ions. The advent of high-throughput techniques including sequencing and saturation mutagenesis has provided large amounts of phenotypic data linked to mutations. However, one of the hurdles has been understanding and quantifying the effects of a particular mutation, and how they translate into a given phenotype. One approach to overcome this is to use robust, accurate and scalable computational methods to understand and correlate structural effects of mutations with disease.
Figures
References
-
- Deng Z., Chuaqui C. & Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J Med Chem 47, 337–344 (2004). - PubMed
-
- Topham C. M., Srinivasan N. & Blundell T. L. Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Protein Eng 10, 7–21 (1997). - PubMed
-
- Capriotti E., Fariselli P. & Casadio R. A neural-network-based method for predicting protein stability changes upon single point mutations. Bioinformatics 20 Suppl 1, i63–68 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
