

Review
doi: 10.1063/1.4865234.
eCollection 2014 Jan.
Topical Review: Molecular reaction and solvation visualized by time-resolved X-ray solution scattering: Structure, dynamics, and their solvent dependence
Affiliations
- PMID: 26798770
- PMCID: PMC4711596
- DOI: 10.1063/1.4865234
Item in Clipboard
Review
Topical Review: Molecular reaction and solvation visualized by time-resolved X-ray solution scattering: Structure, dynamics, and their solvent dependence
Struct Dyn.
.
Abstract
Time-resolved X-ray solution scattering is sensitive to global molecular structure and can track the dynamics of chemical reactions. In this article, we review our recent studies on triiodide ion (I3 (-)) and molecular iodine (I2) in solution. For I3 (-), we elucidated the excitation wavelength-dependent photochemistry and the solvent-dependent ground-state structure. For I2, by combining time-slicing scheme and deconvolution data analysis, we mapped out the progression of geminate recombination and the associated structural change in the solvent cage. With the aid of X-ray free electron lasers, even clearer observation of ultrafast chemical events will be made possible in the near future.
Figures

Schematic representation of the three principal contributions to the X-ray solution
scattering for an I3– ion dissolved in water. The I, O, and H atoms
are colored in purple, red, and white, respectively. Red arrows indicate atomic pairs of
the solute only, while blue and green arrows represent solute–solvent and solvent-only
atomic pairs, respectively.

(a) Candidate structures of of I3– ion in solution. (b) Schematic
of candidate reaction pathways for photodissociation of I3– ion in
solution.

Time-resolved difference X-ray scattering curves of I3– ion in
methanol measured with ((a), (b)) 400 nm and ((c), (d)) 267 nm laser excitation. (a)
Experimental difference scattering curves,
qΔS(q,t), measured with 400 nm laser
excitation (black) and their theoretical fits (red) are shown together. (b) Corresponding
difference radial distribution functions,
rΔS(r,t), obtained by sine-Fourier
transformation of qΔS(q,t) in (a). (c)
Experimental difference scattering curves,
qΔS(q,t), measured with 267 nm laser
excitation (black) and their theoretical fits (red) are shown together. (d) Corresponding
difference radial distribution functions,
rΔS(r,t), obtained by sine-Fourier
transformation of qΔS(q,t) in (c).
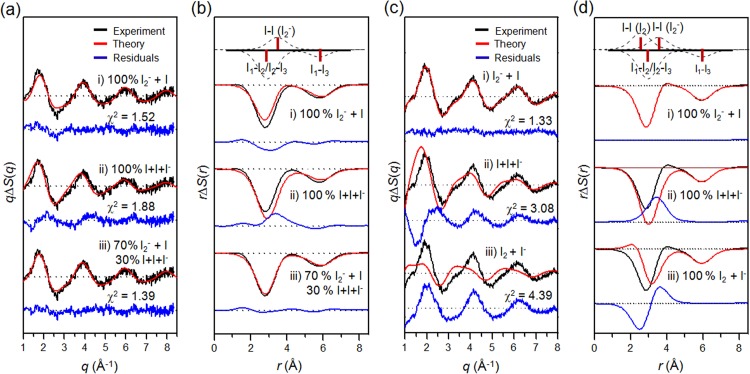
Determination of the reaction pathways of I3– photodissociation in
methanol with ((a), (b)) 400 nm and ((c), (d)) 267 nm laser excitations in ((a), (c))
q-space and ((b), (d)) r-space. (a) Theoretical
difference scattering curve (red) for each candidate pathway is shown together with the
experimental difference scattering curve at 100 ps (black). The model employing only the
two-body dissociation pathway gives much better fit than the models employing the
three-body dissociation and I2 formation pathways, indicating that two-body
dissociation is the dominant reaction pathway with 400 nm laser excitation. (b) Radial
distribution functions, rΔS(r,t), of solute-only term.
Bond distances and their contributions of various I–I pairs are indicated as red bars and
dashed curves, respectively, at the top. With the model employing only two-body
dissociation, the experimental and theoretical RDFs of solute-only term are in good
agreement. (c) The same analysis of (a) for 267 nm laser excitation. The models employing
the two-body and three-body dissociation pathways give similarly good fitting qualities,
indicating the possibility of multiple reaction pathways. The best fit was obtained with a
model employing all three reaction pathways. The optimum ratio of the contributions of
two-body and three-body dissociation was determined to be 7:3. (d) The same analysis of
(b) for 267 nm laser excitation. With the model employing the two-body and three-body
dissociation pathways with the branching ratio of 7:3, the experimental and theoretical
PDFs of solute-only terms are in good agreement.
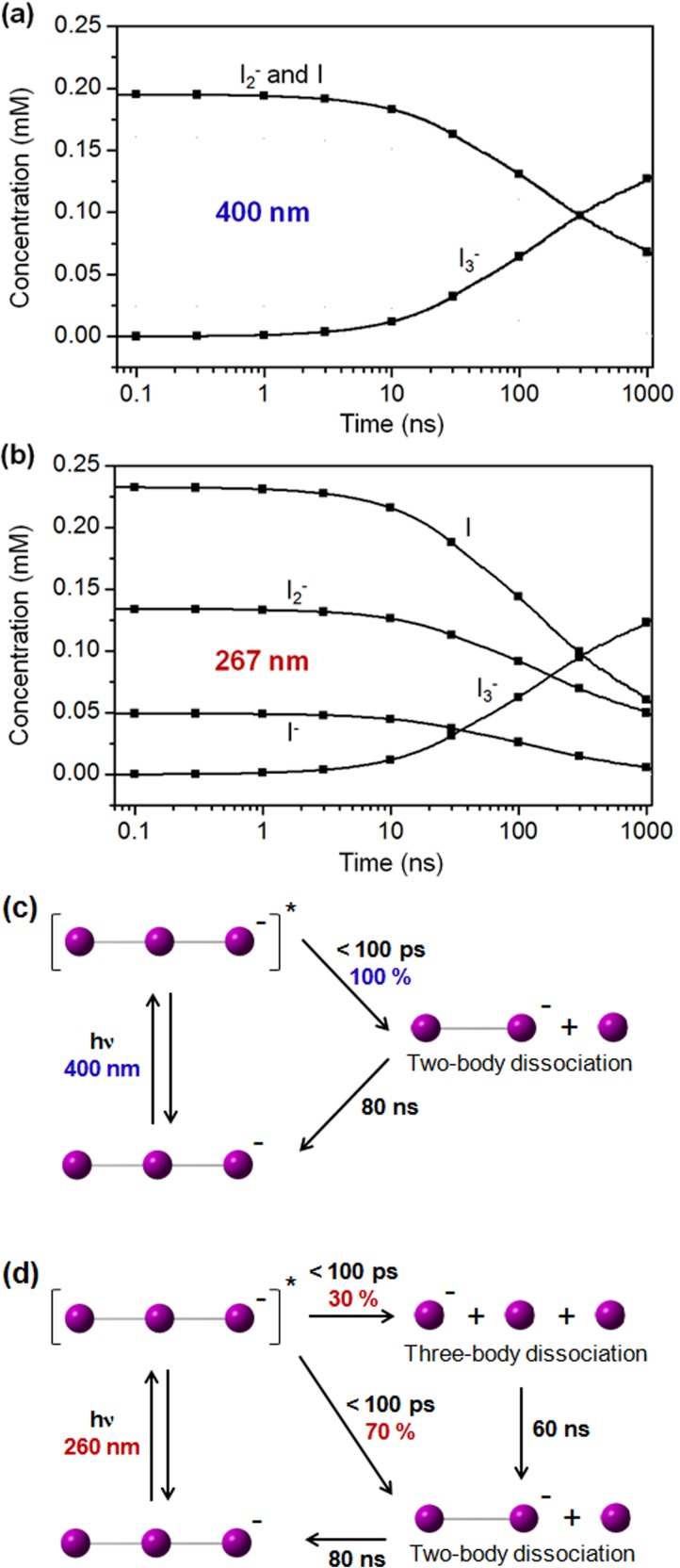
(a) Time-dependent concentration changes of various transient solute species after
photodissociation of I3– ion in methanol with 400 nm excitation. (b)
Time-dependent concentration changes of various transient solute species after
photodissociation of I3– ion in methanol with 267 nm excitation. (c)
Reaction mechanism of I3– photodissociation at 400 nm. (d) Reaction
mechanism of I3– photodissociation at 267 nm.

(a) Atom-atom pairs probed by static X-ray solution scattering. Since X-rays scatter off
from every atom in the solution, the scattering pattern is very complicated. (b)
Scattering intensity of I3– ion extracted from static wide-angle
X-ray solution scattering (black). Scattering patterns from pure solvent and air as well
as the dark response of the detector were subtracted from the scattering pattern of the
solution sample. The theoretical scattering curve (red) does not match the experimental
difference curve due to the unknown background remaining. Therefore, we cannot obtain the
exact structure of I3– ion within a reasonable error range.

Schematic of (a) our experimental approach using TRXSS experiment and (b) data analysis.
Upon irradiation at 400 nm, I3– ion dissociates into
I2– and I, and the temperature of the solution increases. By
taking the difference between scattering patterns measured before and 100 ps after laser
excitation, only the laser-induced changes are extracted with all other background
contributions being eliminated. The structural information can be extracted by the maximum
likelihood estimation using five fitting parameters. Three bond distances of
I3– can be clearly identified, giving the exact structure of
I3– ion in various solvents.

Difference scattering curves measured at 100 ps after photoexcitation for the
I3– photolysis in water, acetonitrile, and methanol solution.
Experimental (black) and theoretical (red) curves using various candidate structures of
I3– ion are compared. Residuals (blue) obtained by subtracting the
theoretical curve from the experimental one are displayed at the bottom. (a) In water,
I3– ion was found to have an asymmetric and bent structure. To
emphasize the fine difference in fitting quality, the residuals shown were multiplied by a
factor of 3. (b) In acetonitrile, I3– ion was found to have a
symmetric and linear structure. (c) In methanol, I3– ion was found
to have an asymmetric and linear structure.
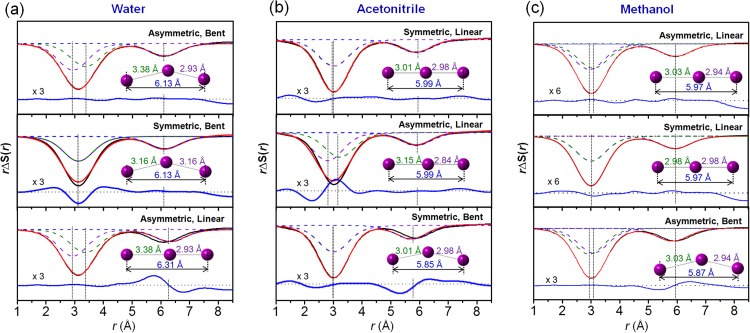
Structure reconstruction of I3– ion based on the extracted bond
distances. The contribution of I3– alone (black solid line) was
extracted. Theoretical curves (red) were generated by a sum of three I–I distances (dashed
lines). The residuals (blue solid line) are displayed at the bottom. (a) In water
solution, the theoretical curve calculated from the asymmetric and bent structure gave the
best fit to the experimental curve (top panel). When one average distance (3.16 Å) instead
of two unequal distances was used, the broad feature in the experimental curve cannot be
matched (middle panel). When a linear and asymmetric structure is used, the sum of two I–I
distances (6.31 Å) do not match the R3 (6.13 Å) determined from the
experimental scattering curve, indicating the bent structure (bottom panel). (b) In
acetonitrile solution, a symmetric and linear structure gave the best fit (top panel). If
two unequal distances (3.15 Å and 2.84 Å) were used, the theoretical curve becomes broader
than the experimental curve (middle panel). When a bent structure was used, the peak at
5.99 Å is shifted to a smaller value, giving a worse fit to the experimental curve
(bottom). (c) In methanol solution, a symmetric and linear structure gave the best fit
(top panel). If two equal distances were used, the theoretical curve becomes slightly
narrower than the experimental curve (middle panel). When a bent structure was used, the
peak at 5.97 Å is shifted to a smaller value, giving a worse fit to the experimental curve
(bottom).

Optimized structures and the relative energies of I3– ion with 34
explicit water molecules forming the first solvation shell by DFT method. The optimized
structure of I3– ion has a broken symmetry (asymmetric, bent) and is
stabilized by 51.2 kJ/mol compared with the linear symmetric one. The elongated iodine
atom has more negative charge than the other iodine atom, resulting in stronger
interaction with surrounding hydrogen atoms of water molecules.
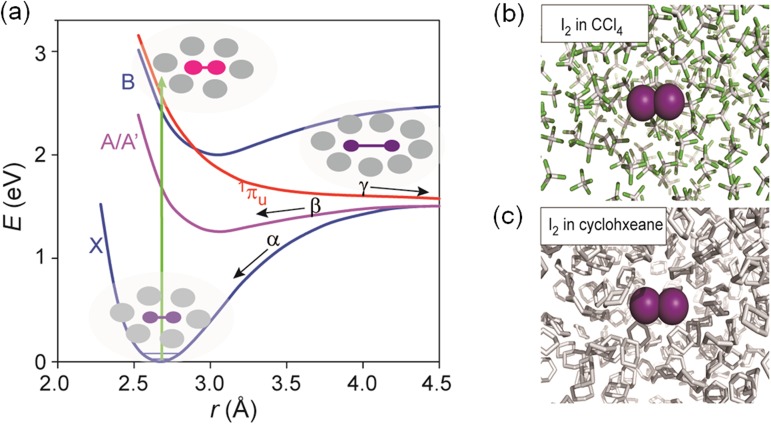
(a) Potential energy surface of I2 in CCl4. Low-lying electronic
states (X, A/A′, B and 1πu) of I2. The states A and A′
are closely spaced and can be viewed as a single electronic state A/A′. The processes α
and β represent geminate recombination of two I atoms in the X and A/A′ states,
respectively. The process γ represents nongeminate recombination through the solvent.
Schematic snapshots of solute-solvent configuration at representative stages are depicted.
(b) MD snapshot of I2 in CCl4. Purple sphere is iodine atom, grey
rod is carbon atom, and green is chloride atom. (c) MD snapshot of I2 in
cyclohexane. Purple sphere is iodine atom, and grey rod is carbon atom.

Schematic of the time-slicing experiment. At a negative time delay (e.g., −30 ps) close
to time zero, the X-ray pulse arrives (effectively) earlier than laser pulse, but the
X-ray pulse, which is much longer in time than the laser pulse, is still present after the
interaction with the laser pulse and thus scattered off the laser-illuminated sample. At
time zero, half of the X-ray pulse probes the laser-illuminated sample. At a positive time
delay, most of the X-ray pulse is scattered off the laser-illuminated sample.
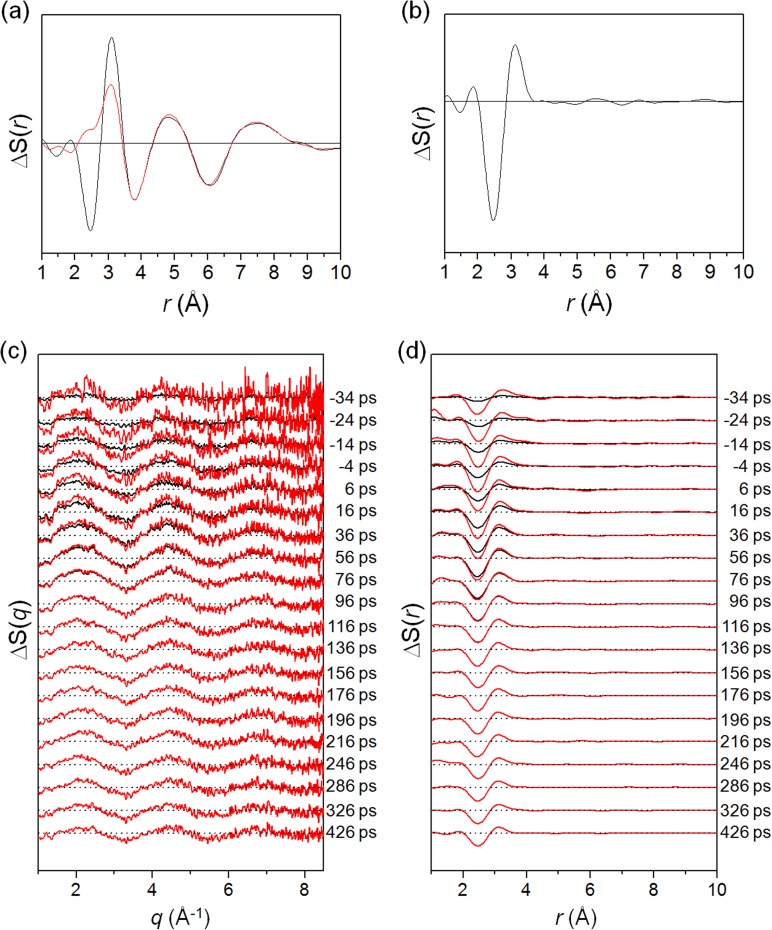
Difference scattering curves for solute and solvent-only contributions. (a)
ΔS(r,426 ps) curve from I2/CCl4
solution (black) and ΔS(r,200 ps)solvent-only
curve from thermally excited CCl4 (red). (b) Solute-related
ΔS(r,426 ps) obtained by subtracting the solvent
contribution from the solution signal. Note the negative peak arising from the depletion
of I2 in the ground (X) state and the positive peak corresponding to the A/A′
state. (c) Solute-related ΔS(q, t) curves with the
solvent contribution eliminated. At early times, only a fraction of the X-ray pulse probes
the laser-triggered molecules and thus the amplitudes of raw difference scattering signals
at early times (black curves) are small. Considering the effect of this partial temporal
overlap, the difference scattering curves at early times were scaled up (red curves)
following the temporal rise of the signal in the form of an error function. (d)
Solute-related ΔS(r, t) curves obtained by Fourier
transform of the curves shown in (c). Note that the depth of the negative peak at 2.6 Å
decreases with time as the geminate recombination progresses and leads to recovery of
I2 in the ground state.

(a) The spectrum of X-ray pulse used in the experiment has a 3% bandwidth and a
characteristic half-Gaussian shape. (b) A scheme for correcting the effect of
polychromatic X-ray spectrum on the difference scattering curve. The polychromaticity of
the X-ray spectrum induces the shift of ΔS(r) along
r-axis (red curve). The black curve is a trial scattering curve
obtained with a monochromatic X-ray beam. When the trial scattering curve is convoluted
with the polychromatic spectrum, a blue curve is generated. By fitting the experimental
data (red curve) with the convoluted trial curve (blue curve) using least-squares
refinement of the trial curve, we can obtain ΔS[r] under
monochromatic conditions.

(a) Concept of deconvolution. If the temporal duration of X-ray pulse is larger or
comparable to the time scale of the process of interest, the dynamic features become
blurred in the experimental data due to the convolution of the sample signal with the
temporal profile of the X-ray pulse. Upper figure shows
r2ΔS(r,t)
with respect to time at r = 3.1 Å. For each r value,
r2ΔS(r,t)
results from the convolution of the sample signal
r2ΔSinst(r,t)
with the X-ray temporal profile Ix-ray(t).
The goal of deconvolution is to reconstruct
r2ΔSinst(r,t)
from
r2ΔS(r,t).
(b) Time-dependent deconvoluted difference scattering curves
r2ΔSinst(r,t)
for I2 in CCl4. (c) The same analysis for I2 in
cyclohexane.

(a)
r2ΔΔSinst(r,t)
for I2 in CCl4 obtained by subtracting
r2ΔSinst(r,426
ps) from
r2ΔSinst(r,t)
to remove the contribution from the A/A′ state and dissociated iodine atoms remaining at
426 ps. (b)
r2ΔΔSinst(r,t)
for I2 in CCl4 obtained by subtracting the contribution from the
population decay of A/A′ (1.2 ns).

(a) Time-dependent I–I distance distribution functions,
r2Sinst(r,t),
of I2 in CCl4. (b) Cross sections of
r2Sinst(r,t)
at the time delays indicated by dotted lines in (a). (c)
⟨r(t)⟩ was calculated from (a) and compared with a
single exponential fit (blue) and a double exponential fit (red). To obtain a satisfactory
fit to the experimental data, a double exponential is necessary with the time constants of
16 ps and 76 ps and a relative amplitude ratio of about 2:1. (d) Time evolution of the I–I
distance distribution function,
r2Sinst(r,t)
(blue, solid line), converted from
ρ(r,t) of the I–I atomic pair
obtained from MD simulation (black, dotted line). The potential energy curve corresponding
to the X state is also shown (red, dashed line). (e) Time dependence of
the average I–I distance, ⟨r⟩, calculated from the I–I distance
distribution function,
r2S(r,t).
Fit of the average distance ⟨r⟩ (blue, solid line) by a double
exponential function,
g(t) = Ar
exp(−t/τ1r) + Br exp(−t/τ2r) + 2.67 Å
(red, dashed line), gives the relaxation times
τ1r = 3 ps and
τ2r = 44 ps. The equilibrium
distance (green, dash-dotted line) is also shown.

(a) Time-dependent I–I distance distribution functions,
r2Sinst(r,t),
of I2 in cyclohexane. (b) Cross sections of
r2Sinst(r,t)
at the time delays indicated by dotted lines in (a). (c)
⟨r(t)⟩ was calculated from (a) and fit by a single
exponential fit (red). The single exponential gives a satisfactory fit to the experimental
data with a time constant of 55 ps.

(a) Experimental
r2ΔSinst(r,t)
curves at large r values corresponding to time-dependent solute–solvent
(mostly I–Cl) distance distribution functions,
r2ΔScage(r,t).
(b) Theoretical time-dependent solute–solvent distance distribution functions,
r2ΔScage(r,t),
based on the experimentally obtained I–I distribution (shown in Figure 17(a)) and the solute-solvent atom–atom pair
distribution functions, g(r), calculated by MD
simulation. (c) Interatomic pair distribution functions between a C atom of the solvent
and an I atom of the solute, gC–I(r). The red
and blue curves are for the solute with the I–I distance of 4.0 Å and 3.1 Å, respectively,
and the black curve is for the solute with the I–I distance of 2.65 Å. (d) Interatomic
pair distribution functions between a Cl atom of the solvent and an I atom of the solute,
gCl–I(r). The red and blue curves are for
the solute with the I–I distance of 4.0 Å and 3.1 Å, respectively, and the black curve is
for the solute with the I–I distance of 2.65 Å. (e) Theoretical solute–solvent distance
distribution function,
r2cageS(r), is
obtained from g(r) – 1 calculated from MD simulation.
(f) Theoretical difference cage term
r2ΔScage(r) is
obtained by subtracting
r2Scage(r) of
I2 in the ground-state configuration from
r2Scage(r) of
I2 with the I–I distance in the range of 2.3 – 4.2 Å. With the decrease of
I–I distance towards the equilibrium distance in the ground state, the width of negative
peak at around 6 Å is narrowed, and positive peak between 7 and 8 Å is shifted to 7 Å.

(a) and (b) Examples of the difference Cl–I pair distribution functions,
ΔgCl–I(r), and the difference C–I pair
distribution functions, ΔgC–I(r), obtained
from the MD simulation. The blue and red curves are for I–I distance changes from 2.65 Å
to 4.0 Å and 2.65 Å to 3.1 Å, respectively. (c) Schematic for the change of the solvation
shell due to the elongation of I–I distance. Dotted circles indicate the first solvation
shell. The interatomic distance shown in this figure is the distance between the I atom of
the solute and the C atom in the solvation shell. Because a CCl4 molecule has
one C atom surrounded by four Cl atoms and the Cl atom scatters much more strongly than
the C atom, the scattering signal is dominated by the I−Cl contribution. Nevertheless,
with the C atom being located at the center of the CCl4 molecule, the I−C
distribution provides a more intuitive picture of the size of the solvation shell.
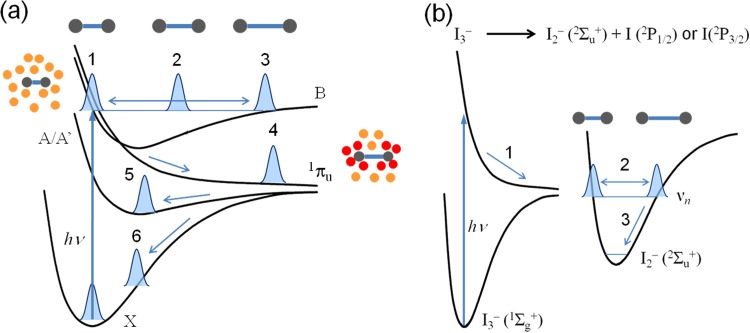
(a) Photodissociation dynamics of I2 in solution phase. Upon photoexcitation
by an optical pulse, coherent vibrational wave packet evolves in the B state to induce the
periodic oscillation of I–I bond length (1, 2, and 3). Subsequently, the excited
population relaxes to a repulsive 1πu state, leading to either
dissociation of I2 (4) or geminate recombination via A/A′ state (5) or hot
ground state (6). (b) Photodissociation dynamics of I3– ion. Upon
photoexcitation of I3– by an optical pulse, one I atom is
dissociated (1), forming coherent vibrational wave packet in the hot ground state
(2Σu+) of I2–. As the coherent
wave packet evolves in the hot ground state of I2– ion, the I–I bond
length of the I2– ion periodically oscillates (2). Subsequently, the
population in the hot ground state of I2– relaxes vibrationally to
the ground state of I2– (3).
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources