

The role of negative selection in protein evolution revealed through the energetics of the native state ensemble
- PMID: 26800099
- PMCID: PMC4811355
- DOI: 10.1002/prot.24989
The role of negative selection in protein evolution revealed through the energetics of the native state ensemble
Erratum in
-
The role of negative selection in protein evolution revealed through the energetics of the native sate ensemble.Proteins. 2018 Dec;86(12):1313. doi: 10.1002/prot.25484. Epub 2018 Mar 1. Proteins. 2018. PMID: 30549116 No abstract available.
Abstract
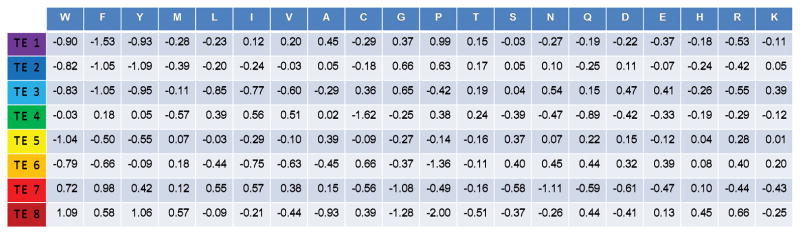
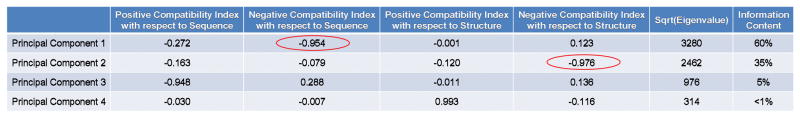
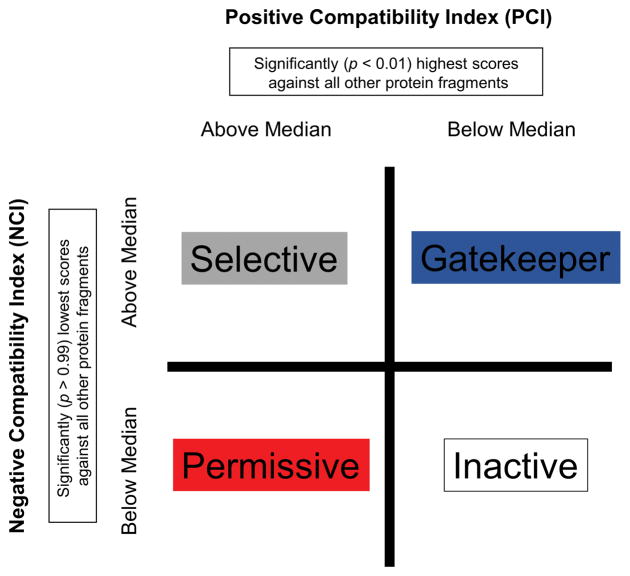
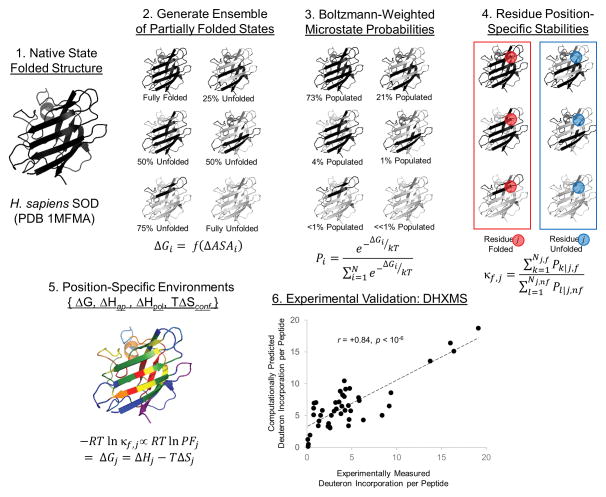
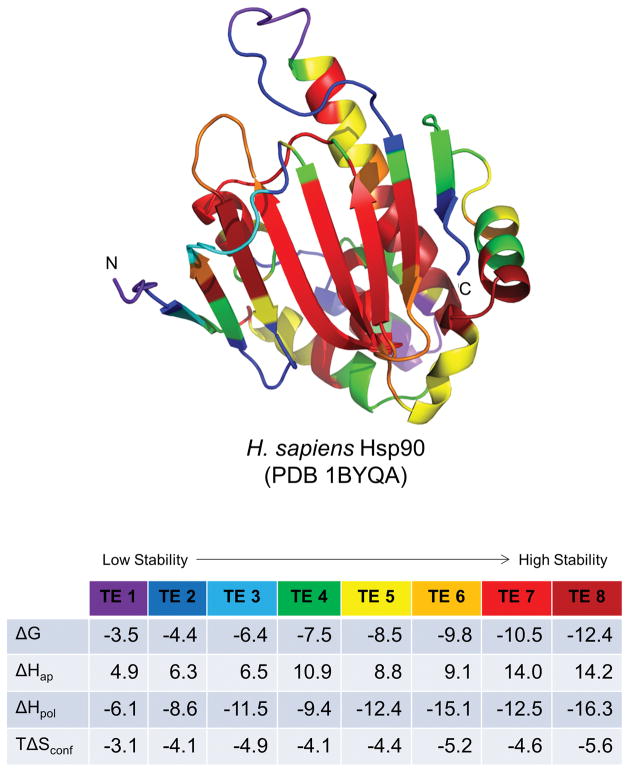
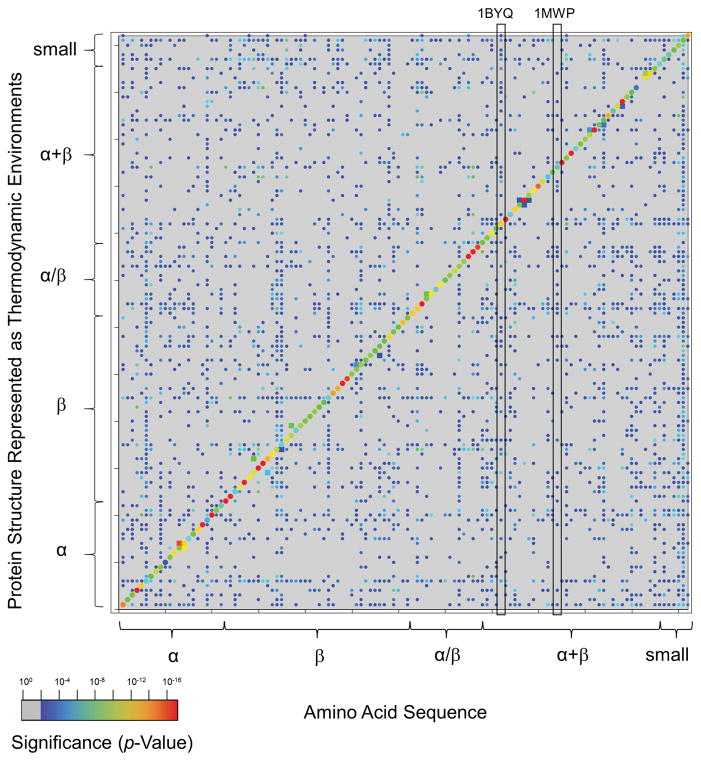
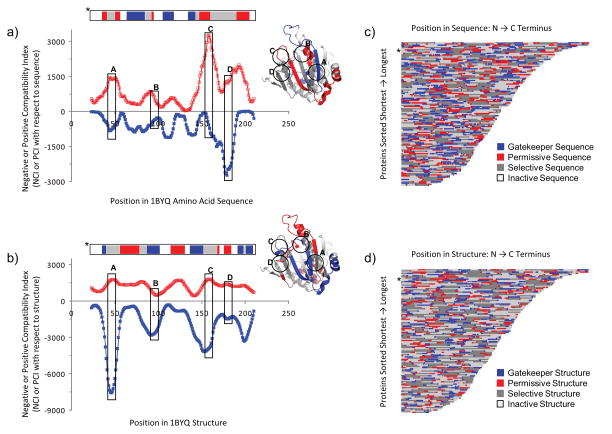
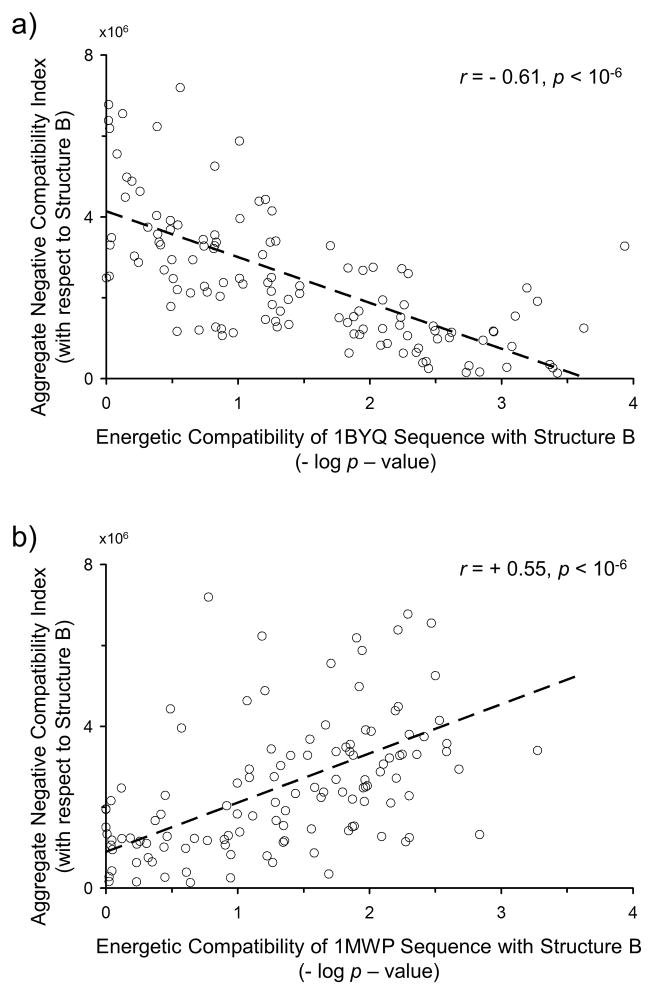
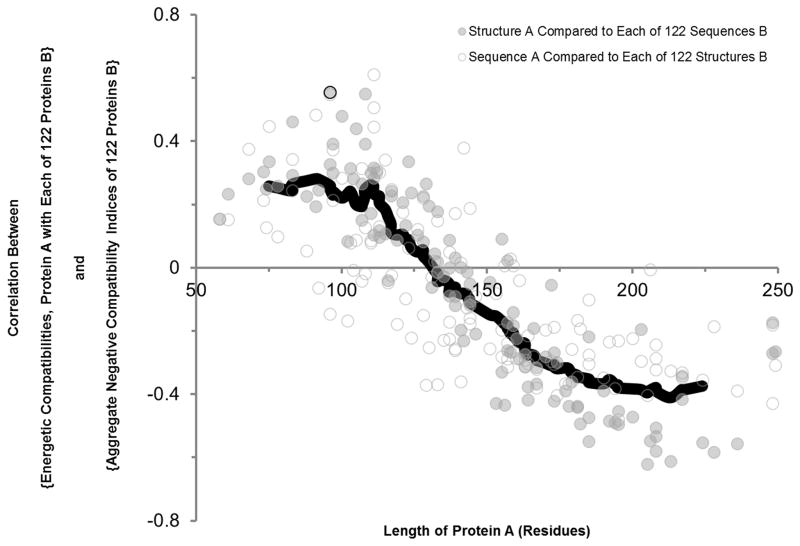
Knowing the determinants of conformational specificity is essential for understanding protein structure, stability, and fold evolution. To address this issue, a novel statistical measure of energetic compatibility between sequence and structure was developed using an experimentally validated model of the energetics of the native state ensemble. This approach successfully matched sequences from a diverse subset of the human proteome to their respective folds. Unexpectedly, significant energetic compatibility between ostensibly unrelated sequences and structures was also observed. Interrogation of these matches revealed a general framework for understanding the origins of conformational specificity within a proteome: specificity is a complex function of both the ability of a sequence to adopt folds other than the native, and ability of a fold to accommodate sequences other than the native. The regional variation in energetic compatibility indicates that the compatibility is dominated by incompatibility of sequence for alternative fold segments, suggesting that evolution of protein sequences has involved substantial negative selection, with certain segments serving as "gatekeepers" that presumably prevent alternative structures. Beyond these global trends, a size dependence exists in the degree to which the energetic compatibility is determined from negative selection, with smaller proteins displaying more negative selection. This partially explains how short sequences can adopt unique folds, despite the higher probability in shorter proteins for small numbers of mutations to increase compatibility with other folds. In providing evolutionary ground rules for the thermodynamic relationship between sequence and fold, this framework imparts valuable insight for rational design of unique folds or fold switches.
Keywords: fold recognition; gapless threading; metamorphic proteins; rational design; thermodynamic environments.
© 2016 Wiley Periodicals, Inc.
Figures
References
-
- Sudarsanam S. Structural diversity of sequentially identical subsequences of proteins: identical octapeptides can have different conformations. Proteins: Structure, Function, and Genetics. 1998;30:228–231. - PubMed
-
- Guo JT, Jaromczyk JW, Xu Y. Analysis of chameleon sequences and their implications in biological processes. Proteins: Structure, Function, and Bioinformatics. 2007;67:548–558. - PubMed
-
- Murzin AG. Metamorphic proteins. Science. 2008;320:1725–1726. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
