

Classification and characterization of human endogenous retroviruses; mosaic forms are common
- PMID: 26800882
- PMCID: PMC4724089
- DOI: 10.1186/s12977-015-0232-y
Classification and characterization of human endogenous retroviruses; mosaic forms are common
Abstract
Background: Human endogenous retroviruses (HERVs) represent the inheritance of ancient germ-line cell infections by exogenous retroviruses and the subsequent transmission of the integrated proviruses to the descendants. ERVs have the same internal structure as exogenous retroviruses. While no replication-competent HERVs have been recognized, some retain up to three of four intact ORFs. HERVs have been classified before, with varying scope and depth, notably in the RepBase/RepeatMasker system. However, existing classifications are bewildering. There is a need for a systematic, unifying and simple classification. We strived for a classification which is traceable to previous classifications and which encompasses HERV variation within a limited number of clades.
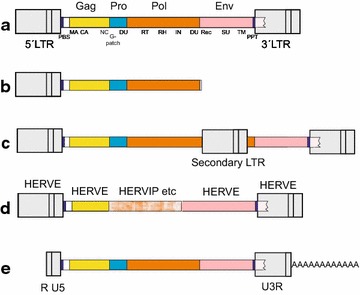
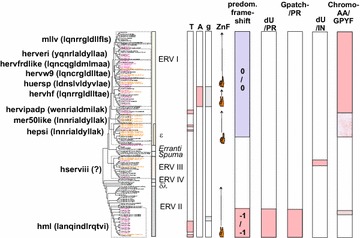
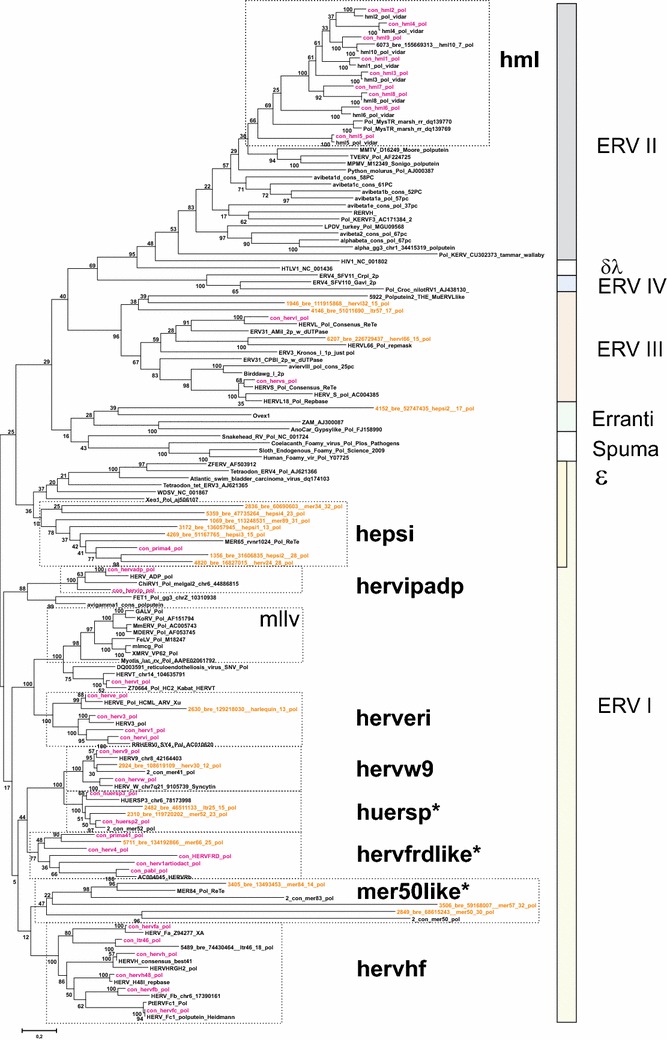
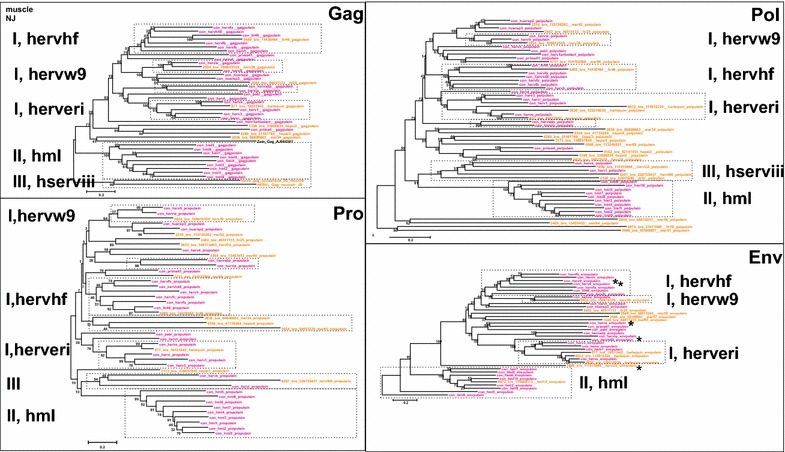
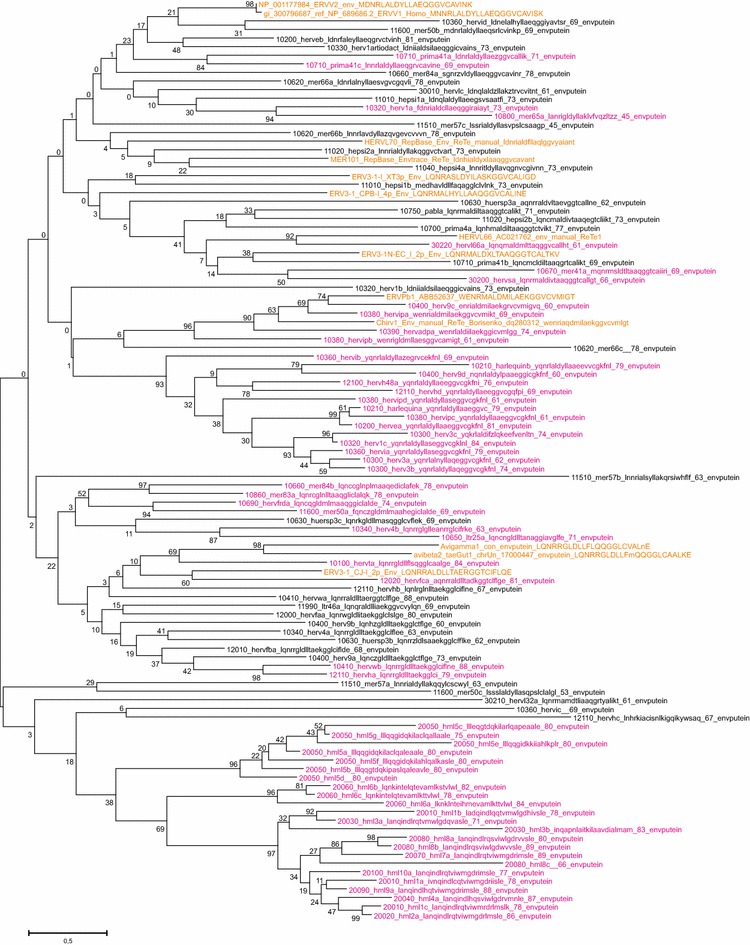
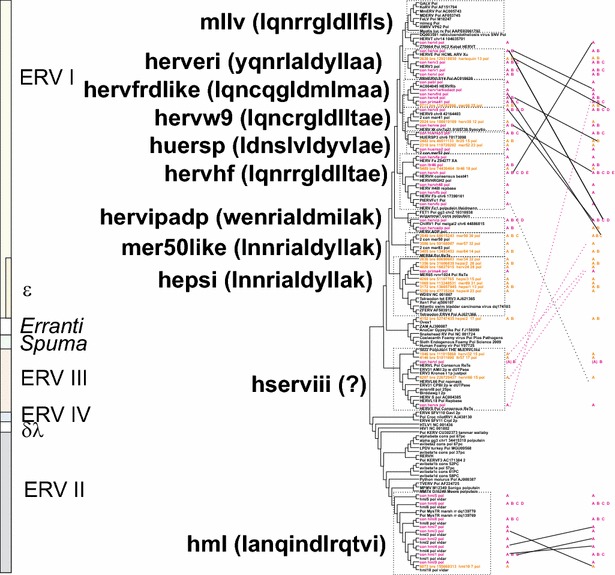
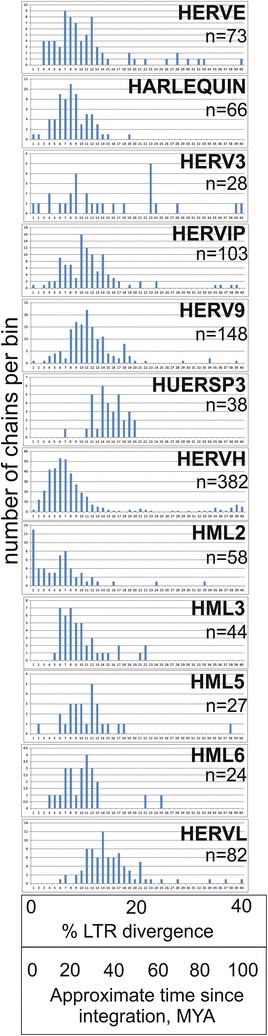
Results: The human genome assembly GRCh 37/hg19 was analyzed with RetroTector, which primarily detects relatively complete Class I and II proviruses. A total of 3173 HERV sequences were identified. The structure of and relations between these proviruses was resolved through a multi-step classification procedure that involved a novel type of similarity image analysis ("Simage") which allowed discrimination of heterogeneous (noncanonical) from homogeneous (canonical) HERVs. Of the 3173 HERVs, 1214 were canonical and segregated into 39 canonical clades (groups), belonging to class I (Gamma- and Epsilon-like), II (Beta-like) and III (Spuma-like). The groups were chosen based on (1) sequence (nucleotide and Pol amino acid), similarity, (2) degree of fit to previously published clades, often from RepBase, and (3) taxonomic markers. The groups fell into 11 supergroups. The 1959 noncanonical HERVs contained 31 additional, less well-defined groups. Simage analysis revealed several types of mosaicism, notably recombination and secondary integration. By comparing flanking sequences, LTRs and completeness of gene structure, we deduced that some noncanonical HERVs proliferated after the recombination event. Groups were further divided into envelope subgroups (altogether 94) based on sequence similarity and characteristic "immunosuppressive domain" motifs. Intra and inter(super)group, as well as intraclass, recombination involving envelope genes ("env snatching") was a common event. LTR divergence indicated that HERV-K(HML2) and HERVFC had the most recent integrations, HERVL and HUERSP3 the oldest.
Conclusions: A comprehensive HERV classification and characterization approach was undertaken. It should be applicable for classification of all ERVs. Recombination was common among HERV ancestors.
Figures

References
-
- Boeke JD, Stoye JP. Retrotransposons, endogenous retroviruses, and the evolution of retroelements. In: Coffin JM, Hughes SH, Varmus HE, editors. retroviruses. New York: Cold Spring Harbor; 1997. - PubMed
-
- Goff SP. Retroviridae: the retroviruses and their replication. In: Knipe D, Howley P, editors. Fields virology 5ed. Philadelpa: Lippincott Williams and Wilkins; 2007.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
