

The Diversity and Molecular Evolution of B-Cell Receptors during Infection
- PMID: 26802217
- PMCID: PMC4839220
- DOI: 10.1093/molbev/msw015
The Diversity and Molecular Evolution of B-Cell Receptors during Infection
Abstract
B-cell receptors (BCRs) are membrane-bound immunoglobulins that recognize and bind foreign proteins (antigens). BCRs are formed through random somatic changes of germline DNA, creating a vast repertoire of unique sequences that enable individuals to recognize a diverse range of antigens. After encountering antigen for the first time, BCRs undergo a process of affinity maturation, whereby cycles of rapid somatic mutation and selection lead to improved antigen binding. This constitutes an accelerated evolutionary process that takes place over days or weeks. Next-generation sequencing of the gene regions that determine BCR binding has begun to reveal the diversity and dynamics of BCR repertoires in unprecedented detail. Although this new type of sequence data has the potential to revolutionize our understanding of infection dynamics, quantitative analysis is complicated by the unique biology and high diversity of BCR sequences. Models and concepts from molecular evolution and phylogenetics that have been applied successfully to rapidly evolving pathogen populations are increasingly being adopted to study BCR diversity and divergence within individuals. However, BCR dynamics may violate key assumptions of many standard evolutionary methods, as they do not descend from a single ancestor, and experience biased mutation. Here, we review the application of evolutionary models to BCR repertoires and discuss the issues we believe need be addressed for this interdisciplinary field to flourish.
Keywords: B-cell receptor; diversity; immunoglobulin; infection.; molecular evolution.
© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
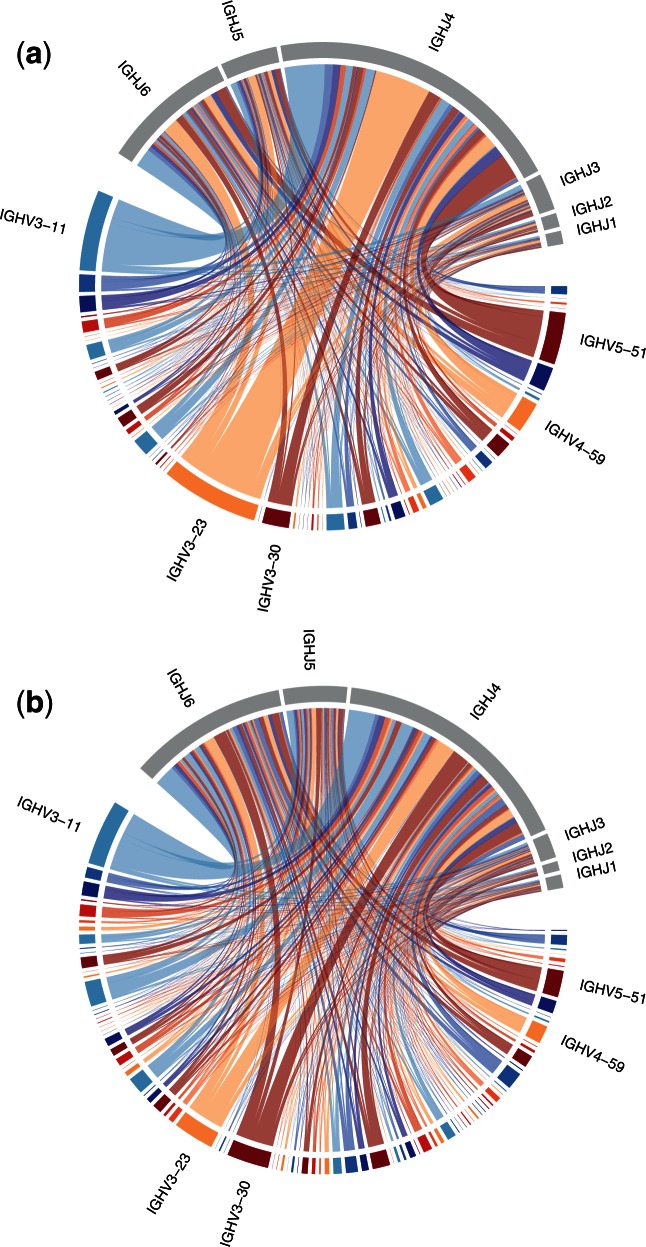
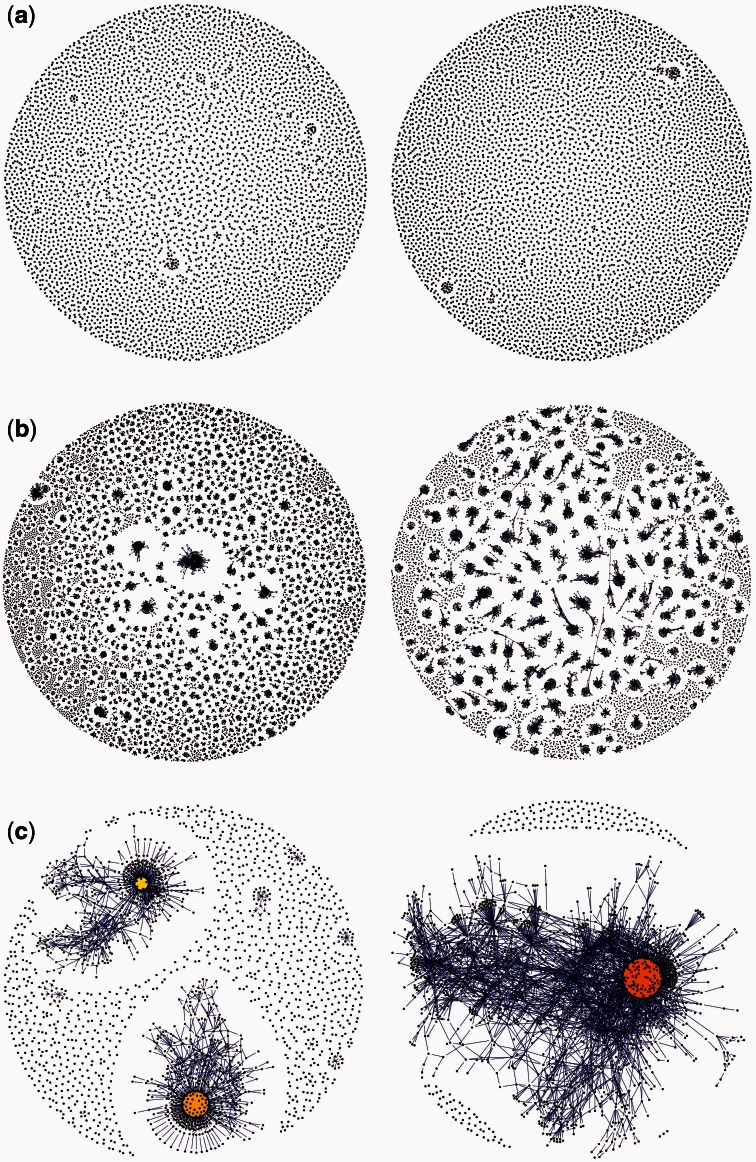

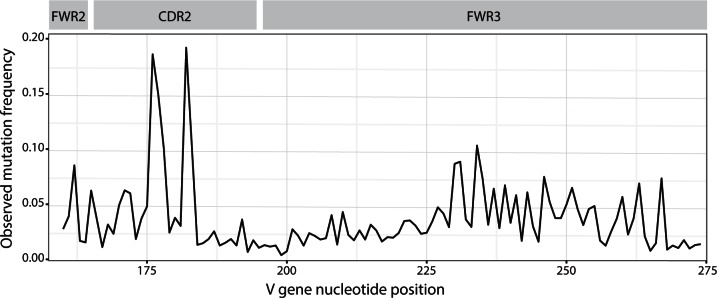
References
-
- Alamyar E, Duroux P, Lefranc M-P, Giudicelli V. 2012. IMGT® tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 882:569–604. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
