

Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study
- PMID: 26803277
- PMCID: PMC6734927
- DOI: 10.1016/S2213-2600(15)00545-7
Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study
Erratum in
-
Corrections.Lancet Respir Med. 2016 Dec;4(12):e57. doi: 10.1016/S2213-2600(16)30386-1. Epub 2016 Nov 22. Lancet Respir Med. 2016. PMID: 27890508 No abstract available.
Abstract
Background: Ivacaftor has been shown to be a safe, effective treatment for cystic fibrosis in patients aged 6 years or older with a CFTR gating mutation. We aimed to assess the safety, pharmacokinetics, and pharmacodynamics of ivacaftor in children aged 2-5 years.
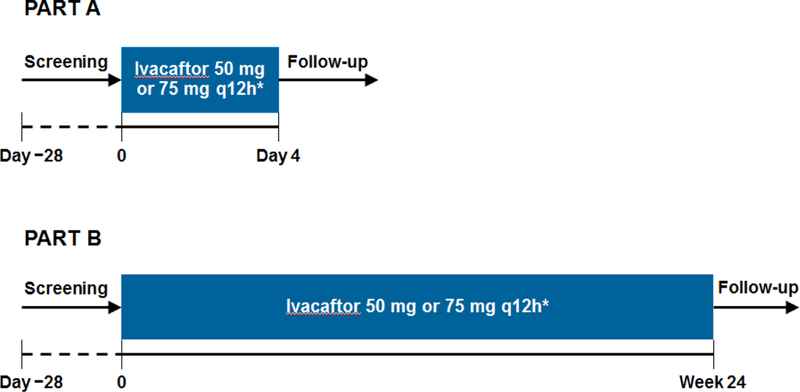
Methods: In the two-part KIWI study, we enrolled children aged 2-5 years weighing 8 kg or more with a confirmed diagnosis of cystic fibrosis and a CFTR gating mutation on at least one allele from 15 hospitals in the USA, UK, and Canada. Participants received oral ivacaftor 50 mg (if bodyweight <14 kg) or 75 mg (if bodyweight ≥14 kg) every 12 h for 4 days in part A (to establish the short-term safety of doses for subsequent assessment in part B), and then for 24 weeks in part B (to assess safety and longer-term pharmacodynamics). Children could participate in both or just one part of the study. Primary outcomes were pharmacokinetics and safety, analysed in all patients who received at least one dose of ivacaftor. Secondary outcomes were absolute change from baseline in sweat chloride concentrations and bodyweight, body-mass index (BMI), and height Z scores, and pharmacokinetic parameter estimation of ivacaftor. This study is registered with ClinicalTrials.gov, number NCT01705145.
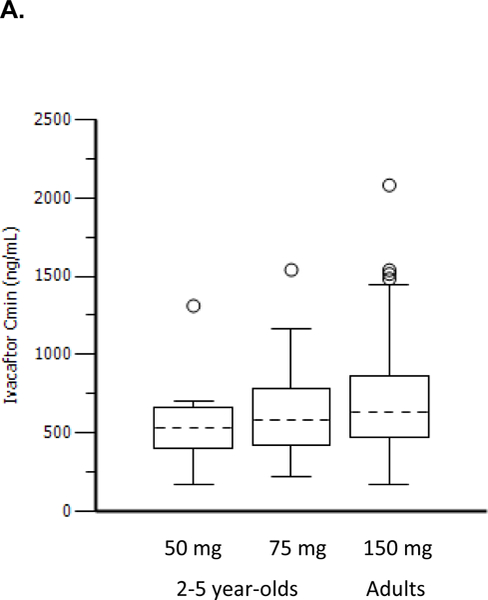
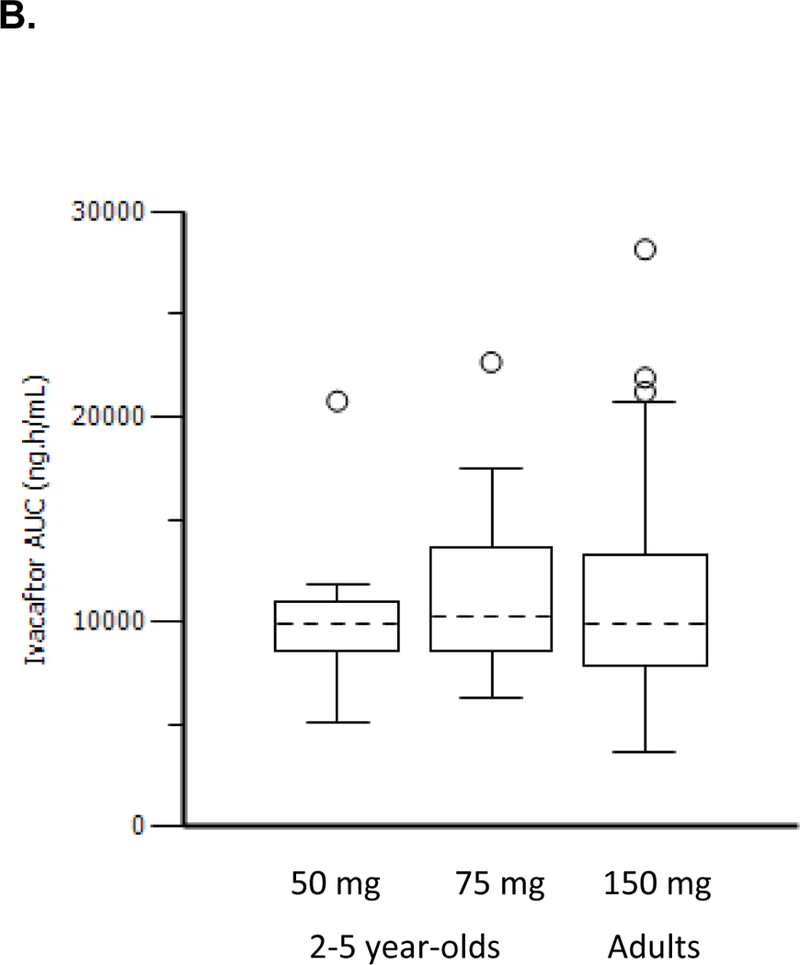
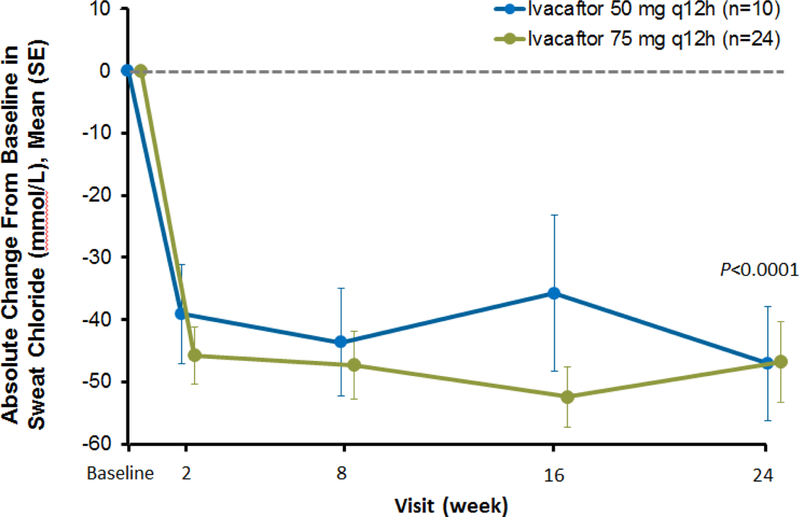
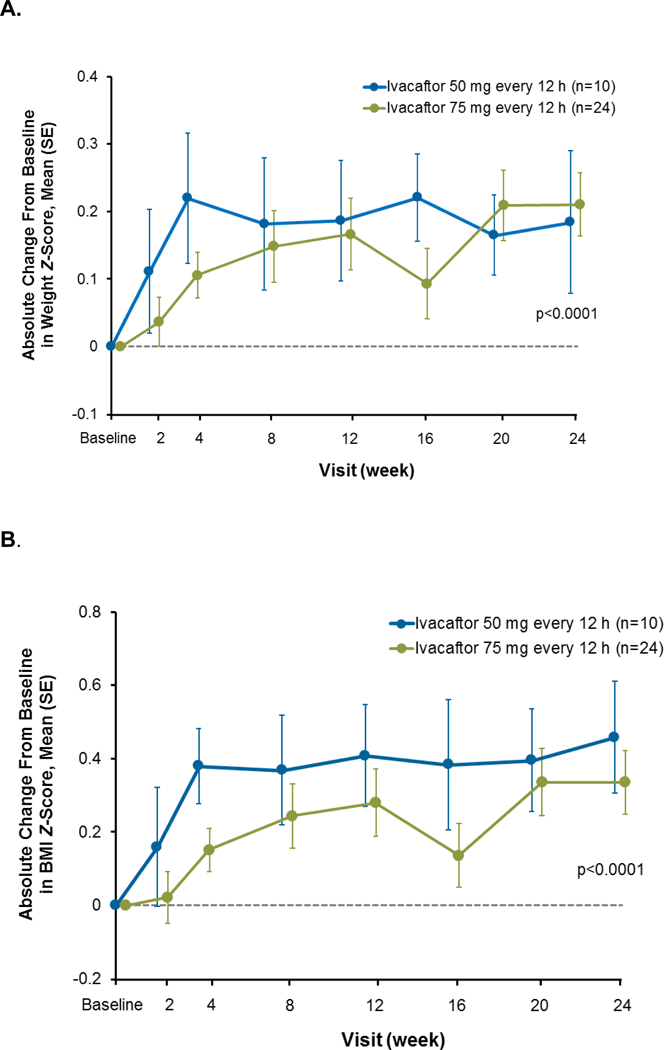
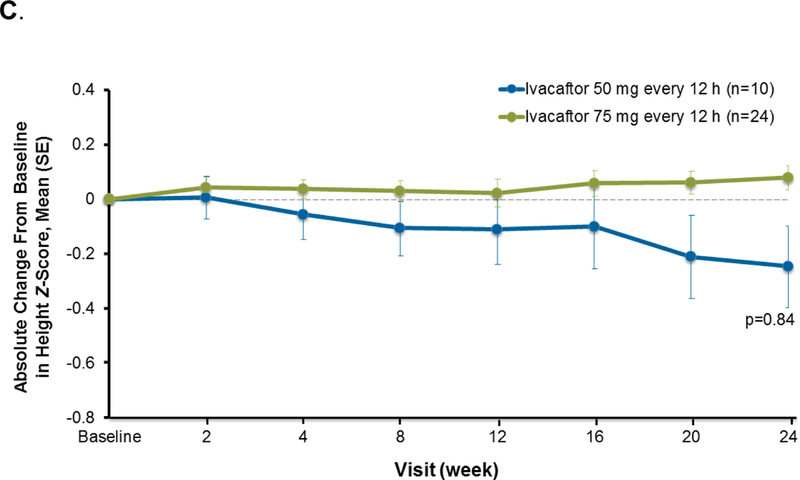
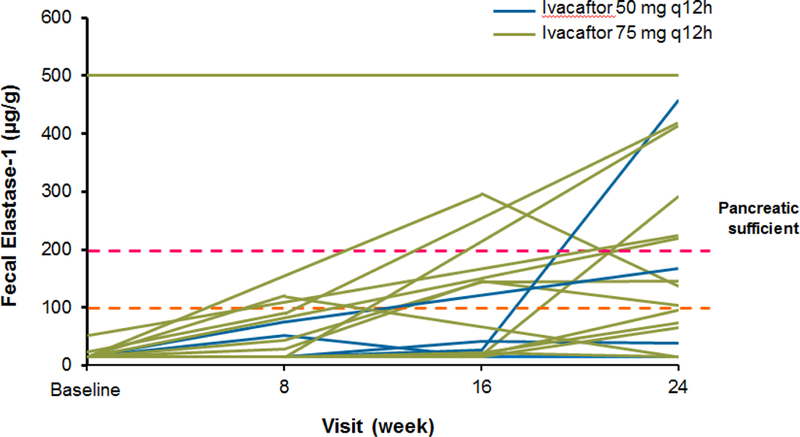
Findings: Between Jan 8, 2013, and March 1, 2013, nine patients were enrolled onto part A of the study, all of whom completed the 4 day treatment period, and eight of whom took part in part B. Between June 28, 2013, and Sept 26, 2013, 34 patients were enrolled in part B, 33 of whom completed the 24 week treatment period. All patients received at least one dose of ivacaftor. Results of ivacaftor pharmacokinetics suggested that exposure was similar to that reported in adults (median Cmin were 536 ng/mL for the 50 mg dose; 580 ng/mL for the 75 mg dose; median ivacaftor AUC values were 9840 ng × h/mL and 10 200 ng × h/mL, respectively). Common adverse events in part B included cough (in 19 [56%] of 34 patients) and vomiting (in ten [29%]). Five (15%) patients had liver function test (LFT) results that were more than eight times higher than the upper limit of normal, four of whom had study drug interrupted, and one of whom had study drug discontinued. Six (18%) of 34 patients had seven serious adverse events; a raised concentration of transaminases was the only serious adverse event regarded as related to ivacaftor and the only adverse event that resulted in study treatment discontinuation. At week 24, in patients for whom we had data, sweat chloride had changed from baseline by a mean of -46·9 mmol/L (SD 26·2, p<0·0001), weight Z score by 0·2 (0·3; p<0·0001), BMI Z score by 0·4 (0·4, p<0·0001), and height Z score by -0·01 (0·3; p=0·84).
Interpretation: Ivacaftor at doses of 50 mg and 75 mg seems to be safe in children aged 2-5 years with cystic fibrosis with a gating mutation followed up for 24 weeks, although the frequency of elevated LFTs suggests that monitoring should be frequent in young children, particularly those with a history of elevated LFTs. Results of an ongoing extension study assessing durability of these effects and longer-term safety are warranted.
Funding: Vertex Pharmaceuticals Incorporated.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
Comment in
-
CFTR modulation for young children with cystic fibrosis.Lancet Respir Med. 2016 Feb;4(2):84-5. doi: 10.1016/S2213-2600(16)00009-6. Epub 2016 Jan 21. Lancet Respir Med. 2016. PMID: 26803278 No abstract available.
-
Partial restoration of pancreatic function in a child with cystic fibrosis.Lancet Respir Med. 2016 May;4(5):e21-2. doi: 10.1016/S2213-2600(16)30032-7. Lancet Respir Med. 2016. PMID: 27304562 No abstract available.
References
-
- Moss R Pathophysiology and therapeutic targets. Johns Hopkins Adv Studies Med 2010; 10: 9–13.
-
- Couper RT, Corey M, Durie PR, Forstner GG, Moore DJ. Longitudinal evaluation of serum trypsinogen measurement in pancreatic-insufficient and pancreatic-sufficient patients with cystic fibrosis. J Pediatr 1995; 127: 408–13. - PubMed
-
- Stick SM, Brennan S, Murray C, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr 2009; 155: 623–8. - PubMed
-
- Li L, Somerset S. Digestive system dysfunction in cystic fibrosis: challenges for nutrition therapy. Dig Liver Dis 2014; 46: 865–74. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
