

Recurrent Muscle Weakness with Rhabdomyolysis, Metabolic Crises, and Cardiac Arrhythmia Due to Bi-allelic TANGO2 Mutations
- PMID: 26805781
- PMCID: PMC4746334
- DOI: 10.1016/j.ajhg.2015.12.008
Recurrent Muscle Weakness with Rhabdomyolysis, Metabolic Crises, and Cardiac Arrhythmia Due to Bi-allelic TANGO2 Mutations
Abstract
The underlying genetic etiology of rhabdomyolysis remains elusive in a significant fraction of individuals presenting with recurrent metabolic crises and muscle weakness. Using exome sequencing, we identified bi-allelic mutations in TANGO2 encoding transport and Golgi organization 2 homolog (Drosophila) in 12 subjects with episodic rhabdomyolysis, hypoglycemia, hyperammonemia, and susceptibility to life-threatening cardiac tachyarrhythmias. A recurrent homozygous c.460G>A (p.Gly154Arg) mutation was found in four unrelated individuals of Hispanic/Latino origin, and a homozygous ∼34 kb deletion affecting exons 3-9 was observed in two families of European ancestry. One individual of mixed Hispanic/European descent was found to be compound heterozygous for c.460G>A (p.Gly154Arg) and the deletion of exons 3-9. Additionally, a homozygous exons 4-6 deletion was identified in a consanguineous Middle Eastern Arab family. No homozygotes have been reported for these changes in control databases. Fibroblasts derived from a subject with the recurrent c.460G>A (p.Gly154Arg) mutation showed evidence of increased endoplasmic reticulum stress and a reduction in Golgi volume density in comparison to control. Our results show that the c.460G>A (p.Gly154Arg) mutation and the exons 3-9 heterozygous deletion in TANGO2 are recurrent pathogenic alleles present in the Latino/Hispanic and European populations, respectively, causing considerable morbidity in the homozygotes in these populations.
Copyright © 2016 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
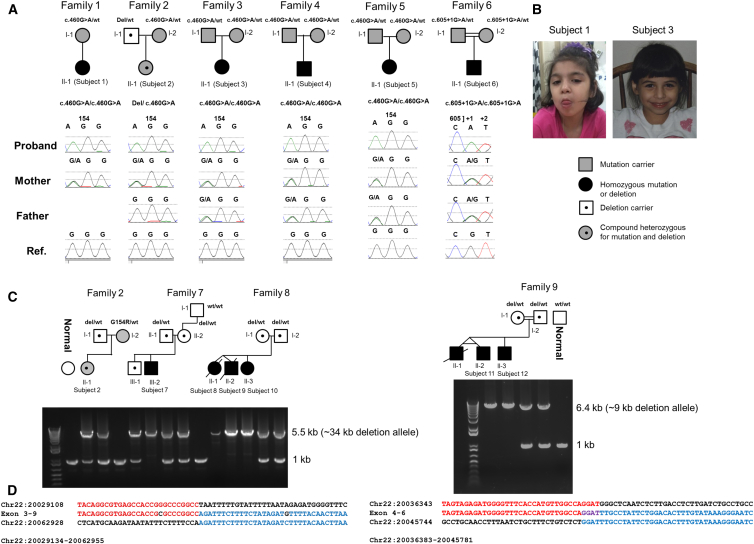
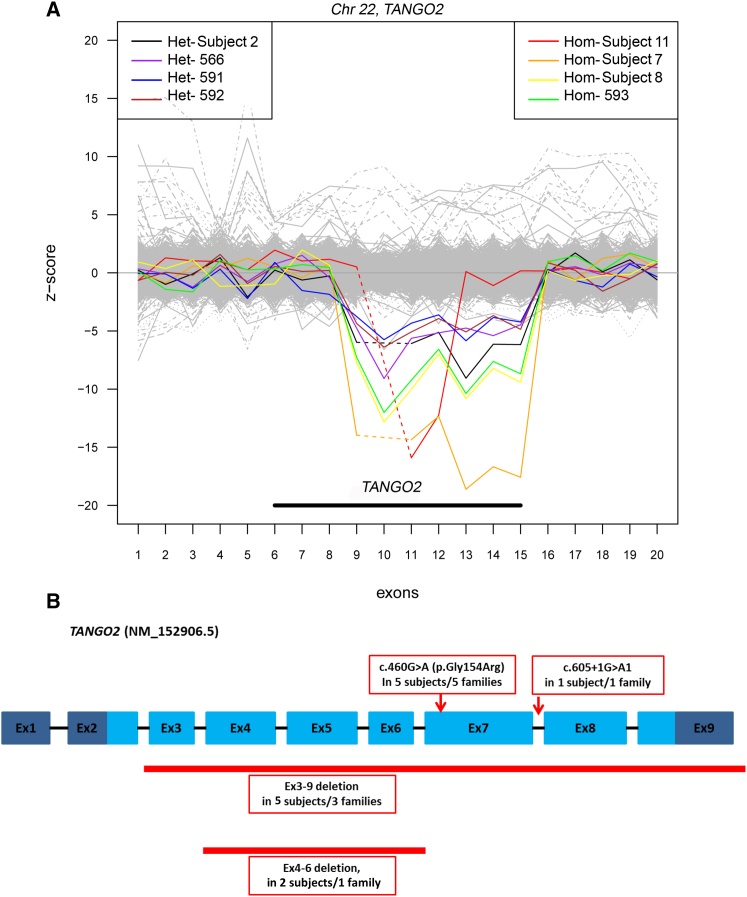
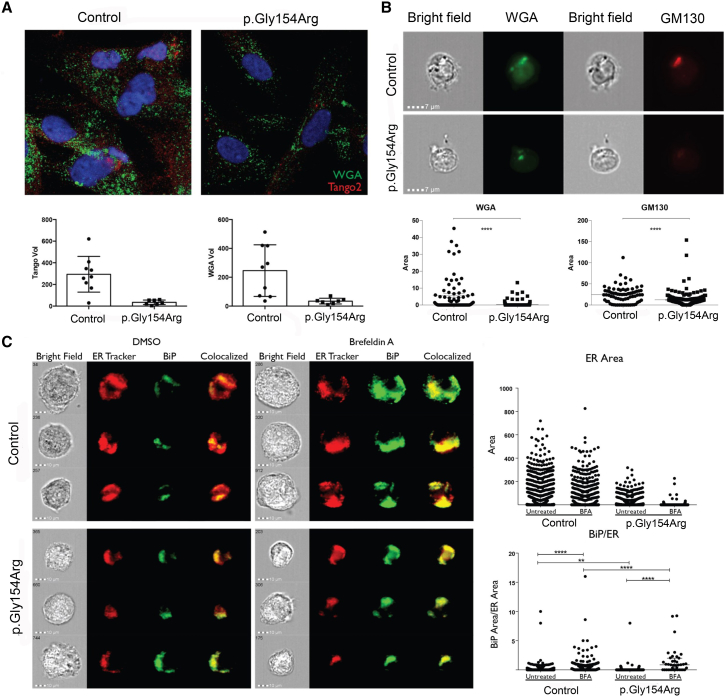
Comment in
-
Is more, "more" when it comes to pacing in heart transplant patients?Heart Rhythm. 2024 Feb;21(2):161-162. doi: 10.1016/j.hrthm.2023.11.002. Epub 2023 Nov 10. Heart Rhythm. 2024. PMID: 37951517 No abstract available.
References
-
- Tonin P., Lewis P., Servidei S., DiMauro S. Metabolic causes of myoglobinuria. Ann. Neurol. 1990;27:181–185. - PubMed
-
- Tyni T., Pihko H. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 1999;88:237–245. - PubMed
-
- Straussberg R., Harel L., Varsano I., Elpeleg O.N., Shamir R., Amir J. Recurrent myoglobinuria as a presenting manifestation of very long chain acyl coenzyme A dehydrogenase deficiency. Pediatrics. 1997;99:894–896. - PubMed
-
- Dlamini N., Voermans N.C., Lillis S., Stewart K., Kamsteeg E.J., Drost G., Quinlivan R., Snoeck M., Norwood F., Radunovic A. Mutations in RYR1 are a common cause of exertional myalgia and rhabdomyolysis. Neuromuscul. Disord. 2013;23:540–548. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
