

De Novo Evolutionary Emergence of a Symmetrical Protein Is Shaped by Folding Constraints
- PMID: 26806127
- PMCID: PMC4735018
- DOI: 10.1016/j.cell.2015.12.024
De Novo Evolutionary Emergence of a Symmetrical Protein Is Shaped by Folding Constraints
Abstract
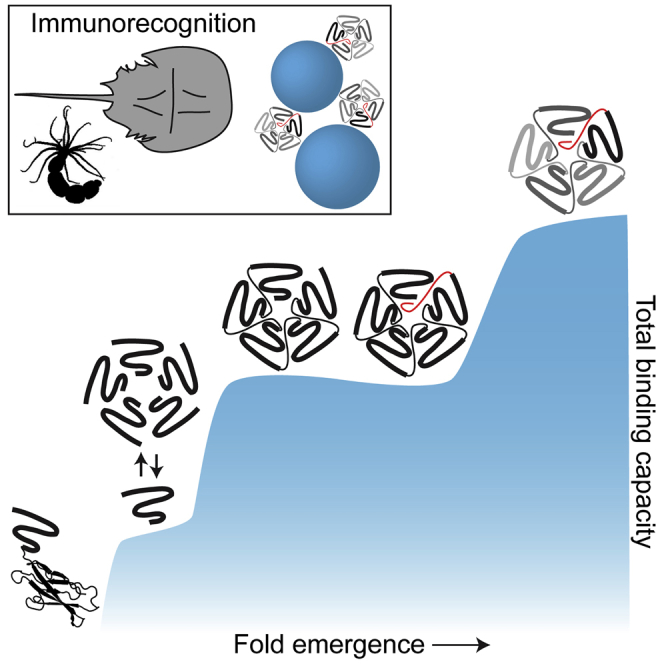
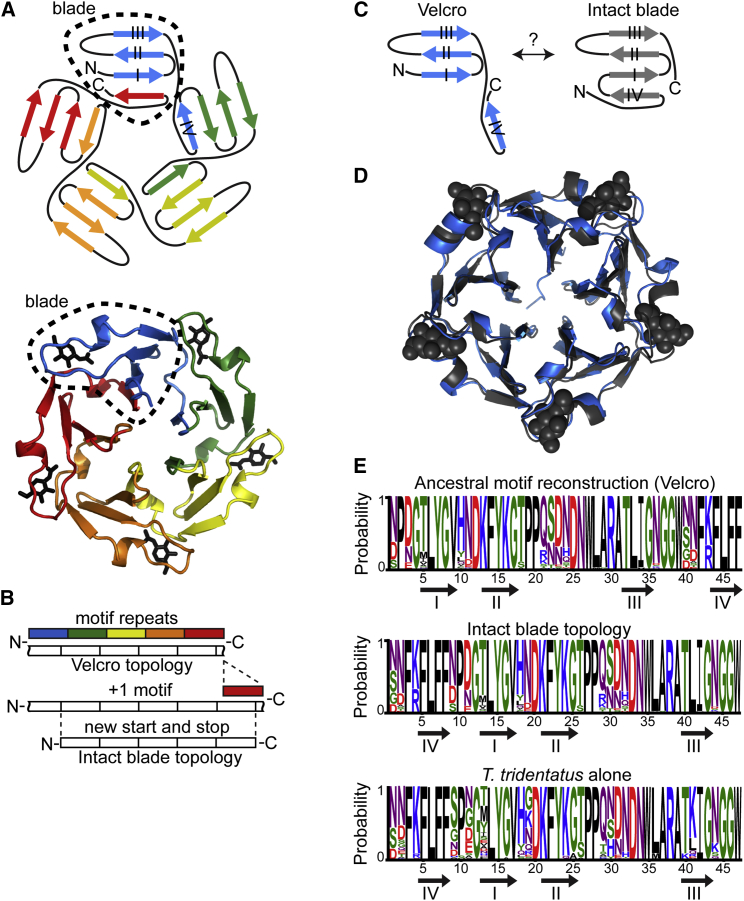
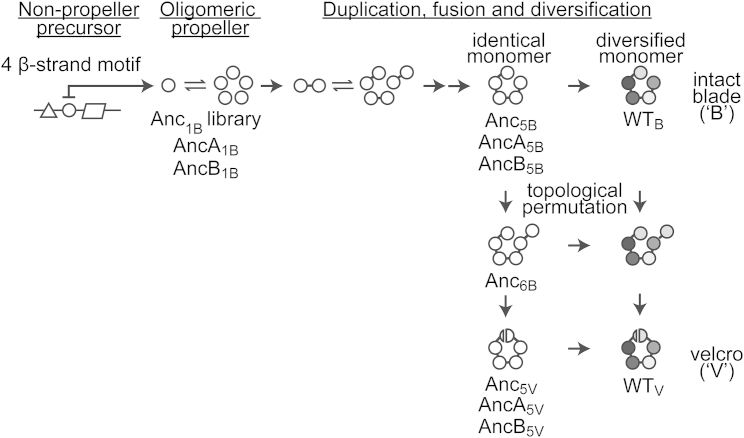
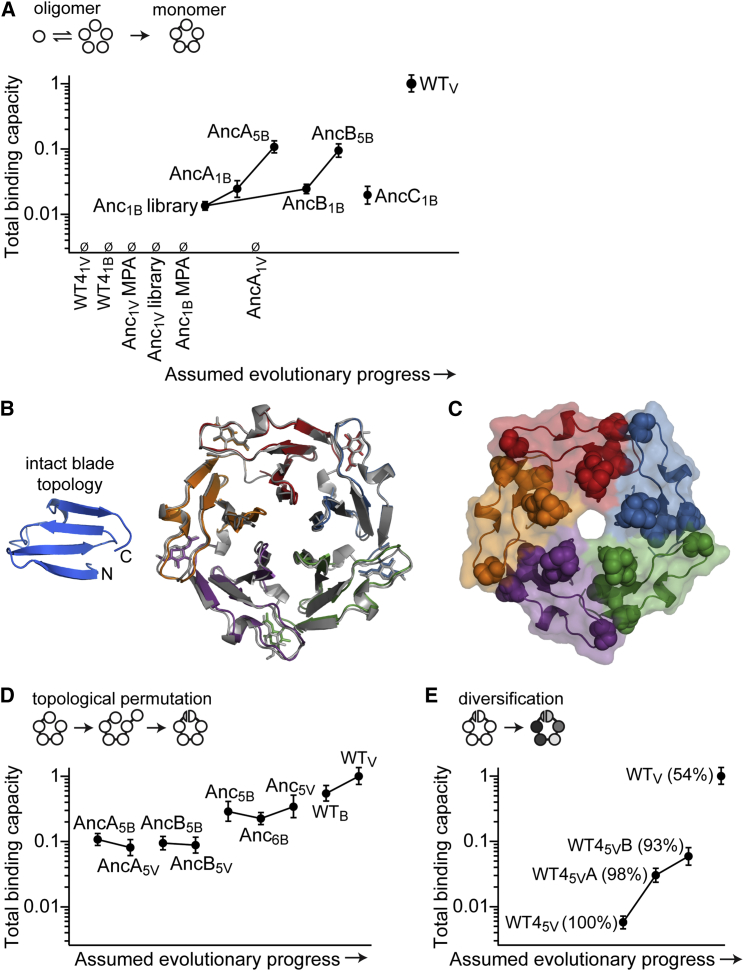
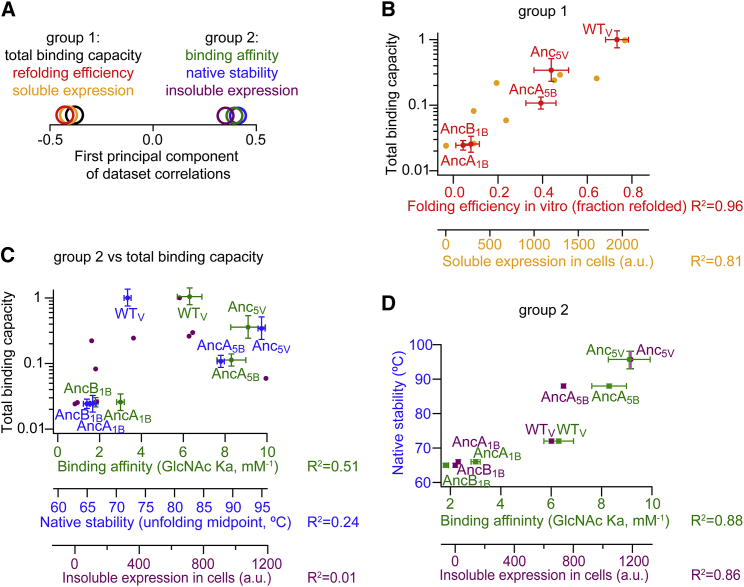
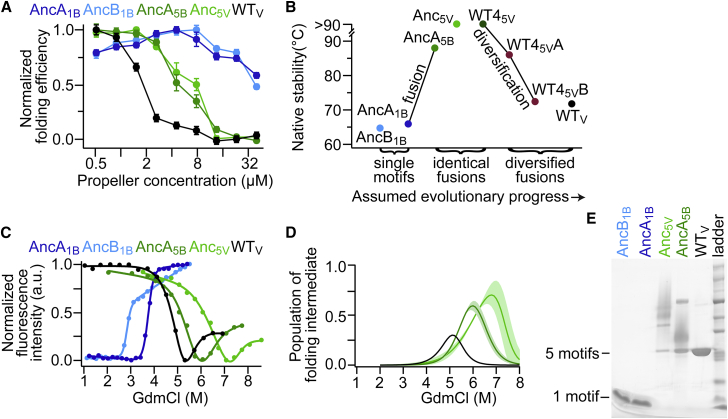
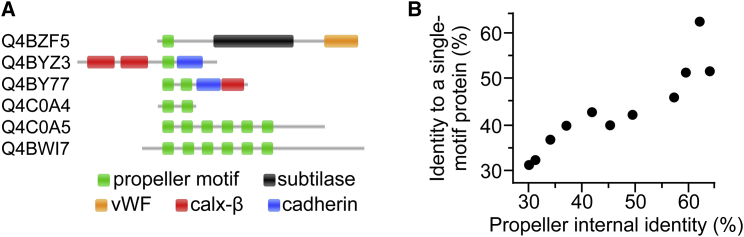
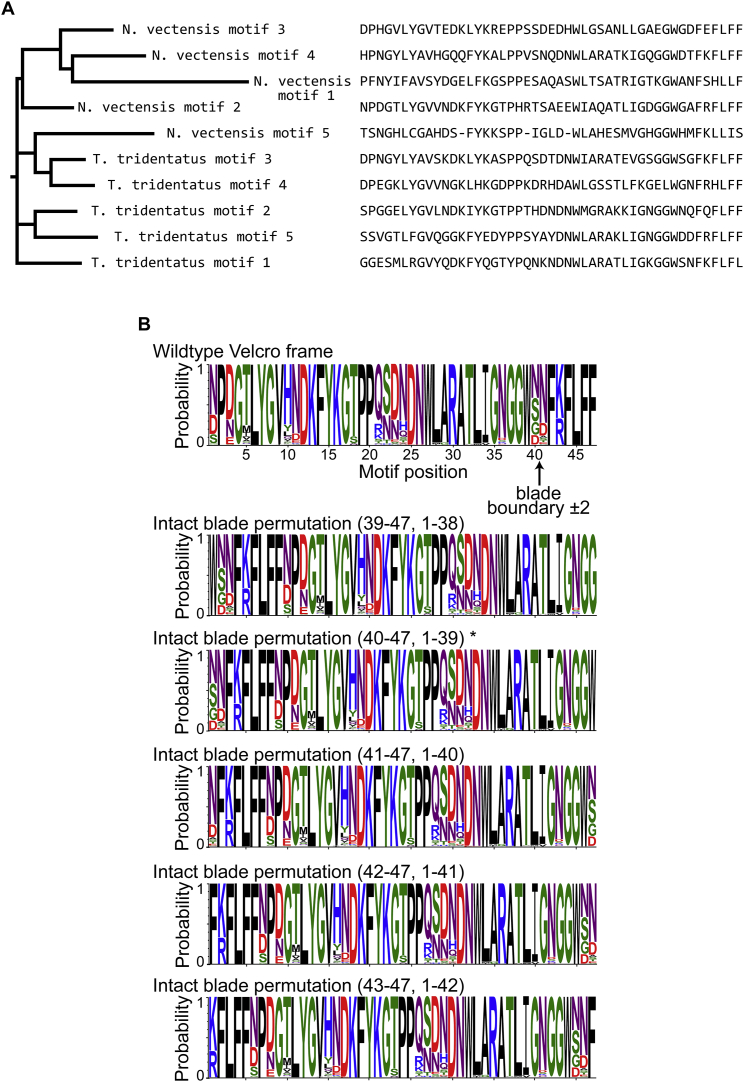
Molecular evolution has focused on the divergence of molecular functions, yet we know little about how structurally distinct protein folds emerge de novo. We characterized the evolutionary trajectories and selection forces underlying emergence of β-propeller proteins, a globular and symmetric fold group with diverse functions. The identification of short propeller-like motifs (<50 amino acids) in natural genomes indicated that they expanded via tandem duplications to form extant propellers. We phylogenetically reconstructed 47-residue ancestral motifs that form five-bladed lectin propellers via oligomeric assembly. We demonstrate a functional trajectory of tandem duplications of these motifs leading to monomeric lectins. Foldability, i.e., higher efficiency of folding, was the main parameter leading to improved functionality along the entire evolutionary trajectory. However, folding constraints changed along the trajectory: initially, conflicts between monomer folding and oligomer assembly dominated, whereas subsequently, upon tandem duplication, tradeoffs between monomer stability and foldability took precedence.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
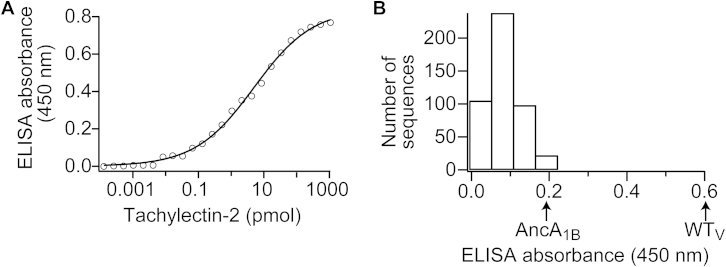
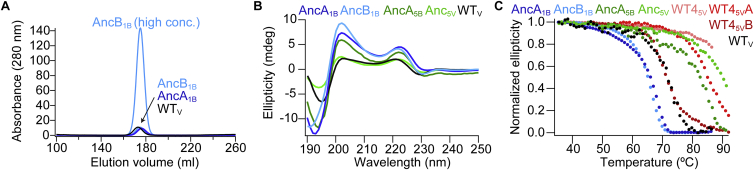
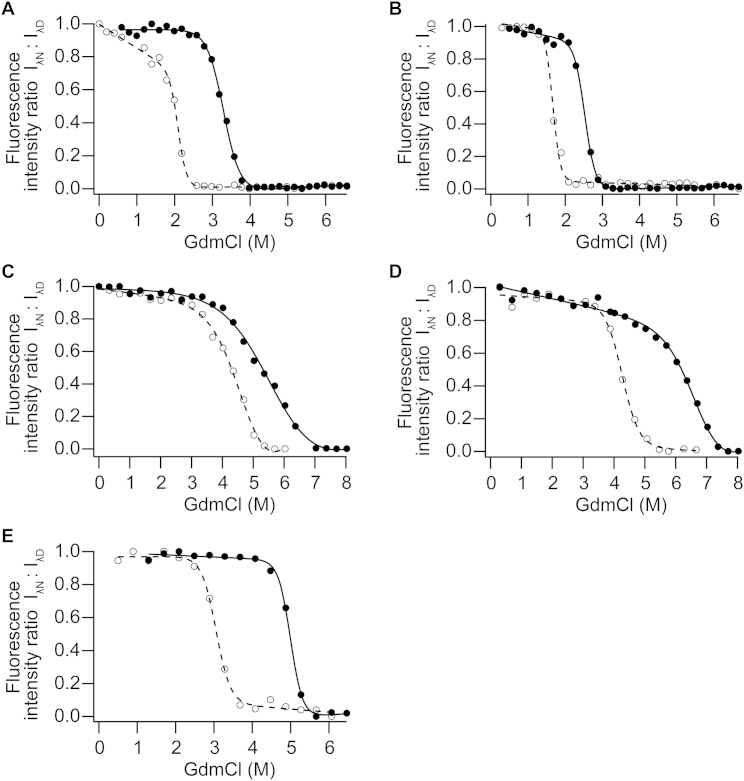
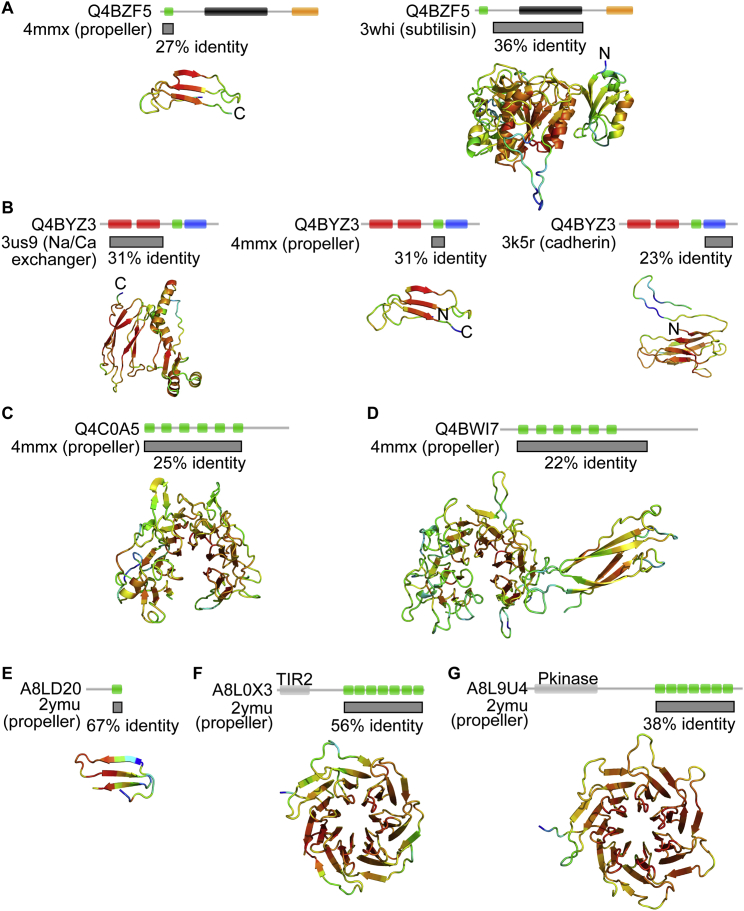
Comment in
-
How to Build a Complex, Functional Propeller Protein, From Parts.Trends Biochem Sci. 2016 Apr;41(4):290-292. doi: 10.1016/j.tibs.2016.02.010. Epub 2016 Mar 9. Trends Biochem Sci. 2016. PMID: 26971075 Free PMC article.
References
-
- Afriat-Jurnou L., Jackson C.J., Tawfik D.S. Reconstructing a missing link in the evolution of a recently diverged phosphotriesterase by active-site loop remodeling. Biochemistry. 2012;51:6047–6055. - PubMed
-
- Balaji S. Internal symmetry in protein structures: prevalence, functional relevance and evolution. Curr. Opin. Struct. Biol. 2015;32:156–166. - PubMed
-
- Bar-Rogovsky H., Stern A., Penn O., Kobl I., Pupko T., Tawfik D.S. Assessing the prediction fidelity of ancestral reconstruction by a library approach. Protein Eng. Des. Sel. 2015;28:507–518. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
