

Prediction of donor splice sites using random forest with a new sequence encoding approach
- PMID: 26807151
- PMCID: PMC4724119
- DOI: 10.1186/s13040-016-0086-4
Prediction of donor splice sites using random forest with a new sequence encoding approach
Abstract
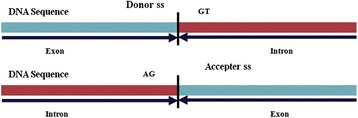
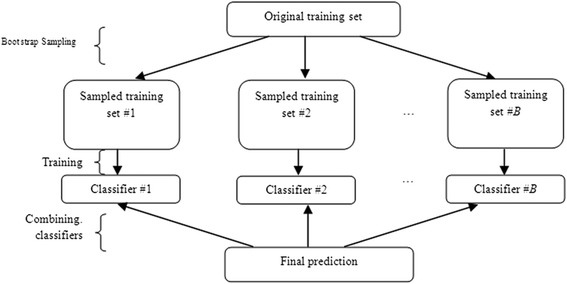
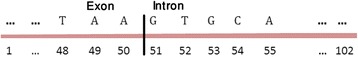
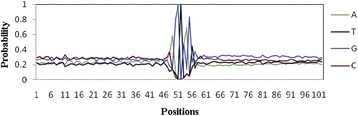
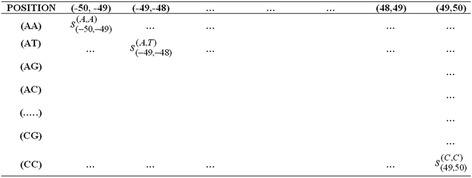
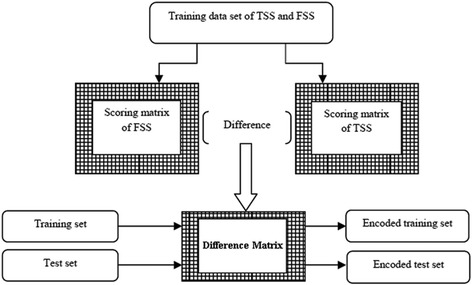
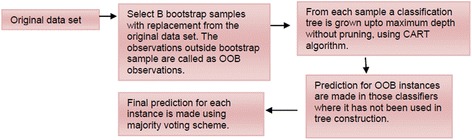
Background: Detection of splice sites plays a key role for predicting the gene structure and thus development of efficient analytical methods for splice site prediction is vital. This paper presents a novel sequence encoding approach based on the adjacent di-nucleotide dependencies in which the donor splice site motifs are encoded into numeric vectors. The encoded vectors are then used as input in Random Forest (RF), Support Vector Machines (SVM) and Artificial Neural Network (ANN), Bagging, Boosting, Logistic regression, kNN and Naïve Bayes classifiers for prediction of donor splice sites.
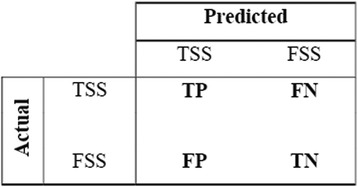
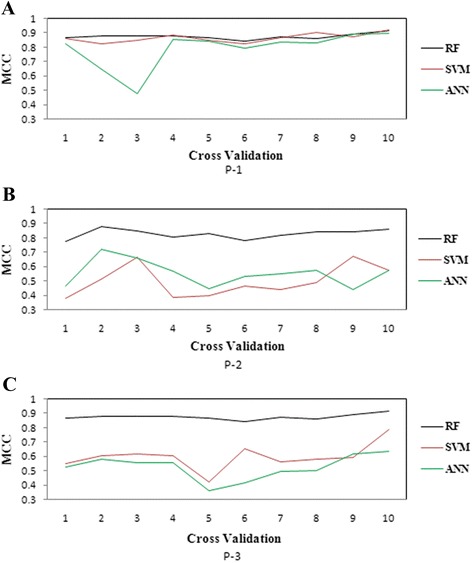
Results: The performance of the proposed approach is evaluated on the donor splice site sequence data of Homo sapiens, collected from Homo Sapiens Splice Sites Dataset (HS3D). The results showed that RF outperformed all the considered classifiers. Besides, RF achieved higher prediction accuracy than the existing methods viz., MEM, MDD, WMM, MM1, NNSplice and SpliceView, while compared using an independent test dataset.
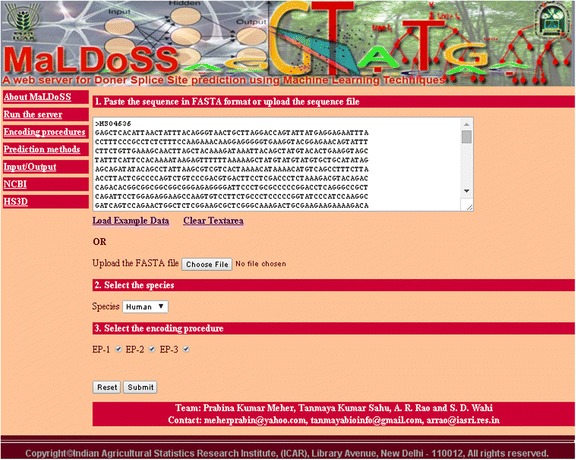
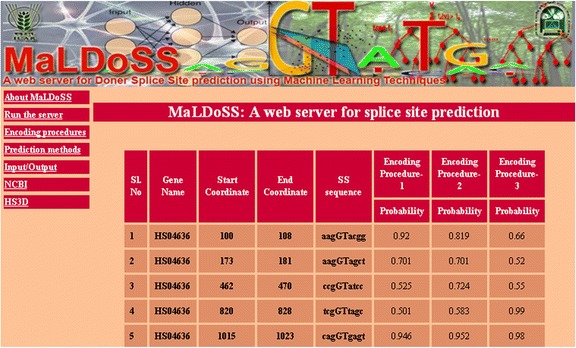
Conclusion: Based on the proposed approach, we have developed an online prediction server (MaLDoSS) to help the biological community in predicting the donor splice sites. The server is made freely available at http://cabgrid.res.in:8080/maldoss. Due to computational feasibility and high prediction accuracy, the proposed approach is believed to help in predicting the eukaryotic gene structure.
Keywords: Computational feasibility; Di-nucleotide association; Machine learning; PWM.
Figures
References
-
- Rajapakse J, CaH LS. Markov encoding for detecting signals in genomic sequences. IEEE Trans Comput. Biol Bioinformatics. 2005;2(2):131–142. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
