

Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements
- PMID: 26807575
- PMCID: PMC4726740
- DOI: 10.1371/journal.pntd.0004402
Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements
Abstract
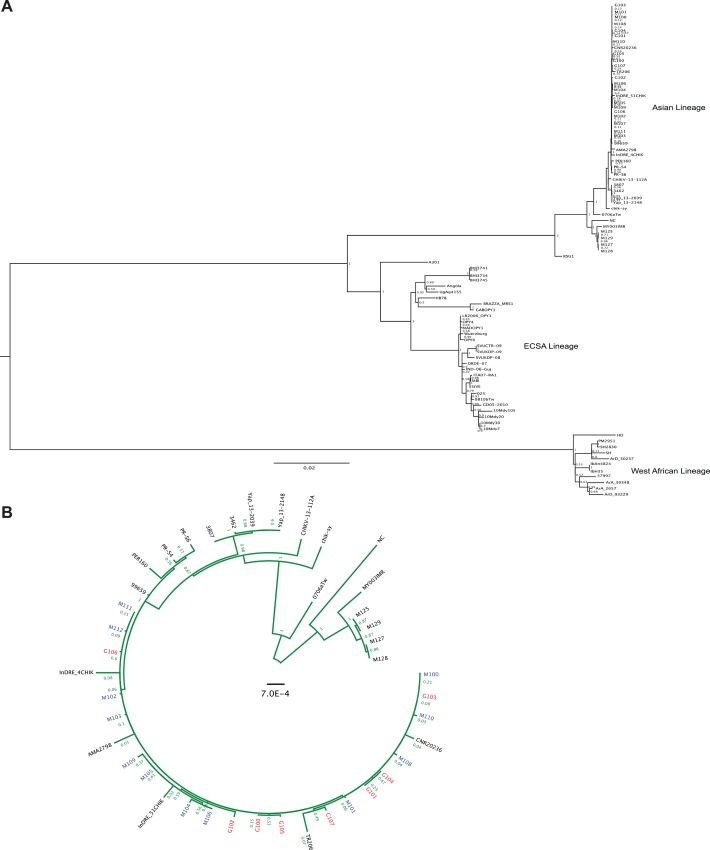
Background: Chikungunya virus (CHIKV), an alphavirus and member of the Togaviridae family, is capable of causing severe febrile disease in humans. In December of 2013 the Asian Lineage of CHIKV spread from the Old World to the Americas, spreading rapidly throughout the New World. Given this new emergence in naïve populations we studied the viral genetic diversity present in infected individuals to understand how CHIKV may have evolved during this continuing outbreak.
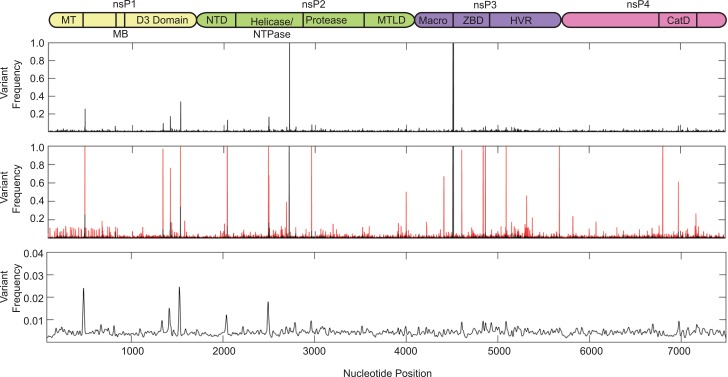
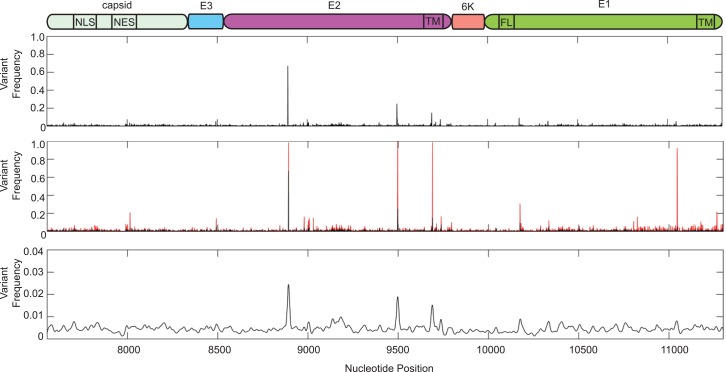
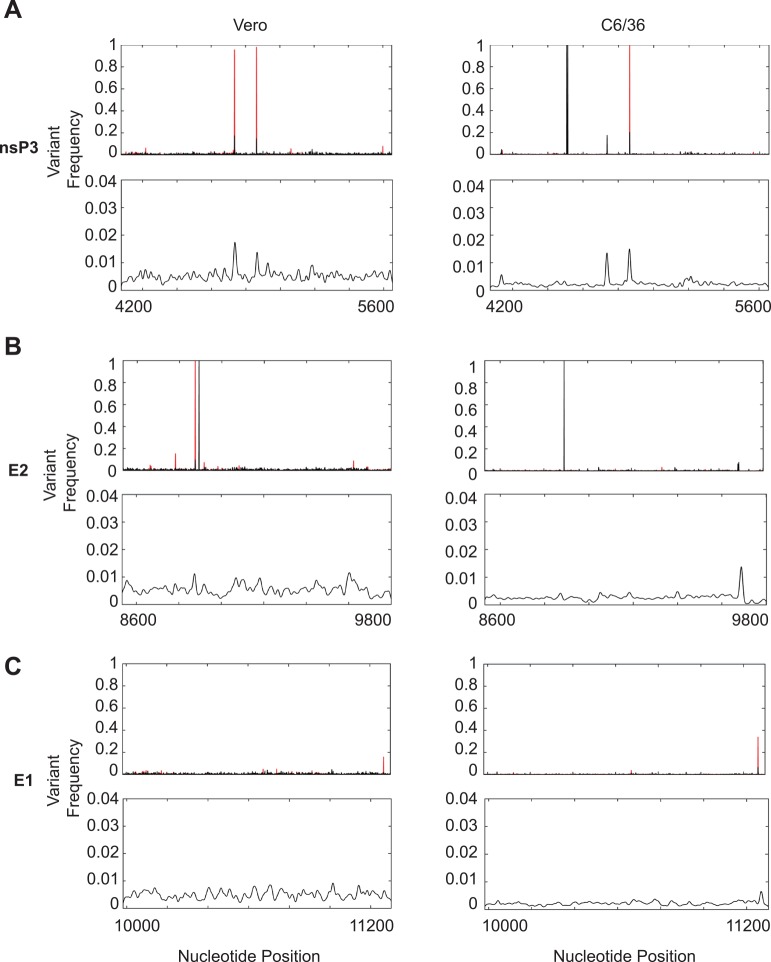
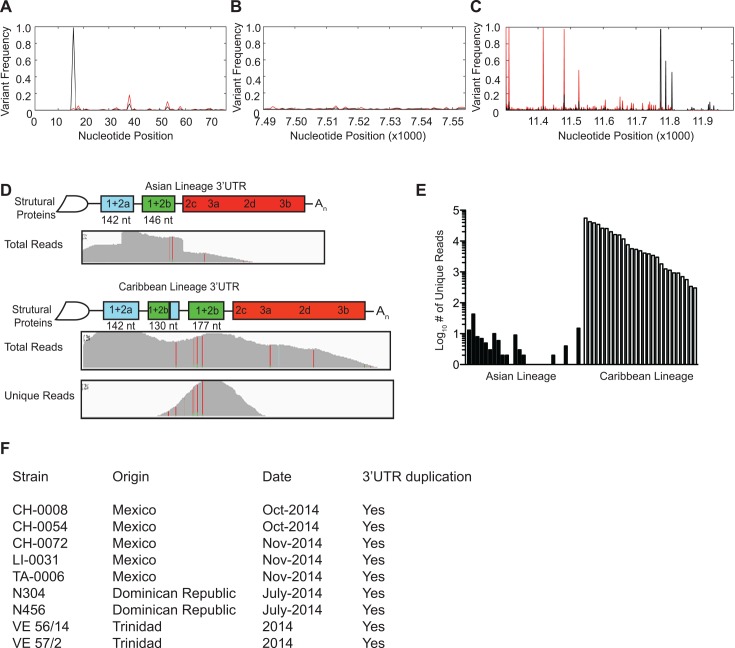
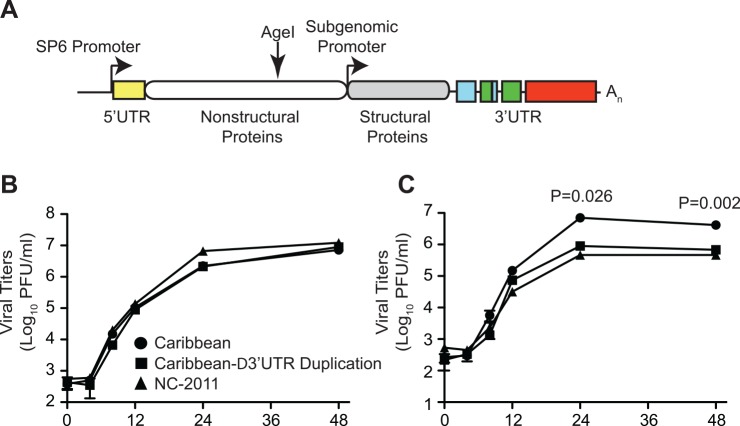
Methodology/principle findings: We used deep-sequencing technologies coupled with well-established bioinformatics pipelines to characterize the minority variants and diversity present in CHIKV infected individuals from Guadeloupe and Martinique, two islands in the center of the epidemic. We observed changes in the consensus sequence as well as a diverse range of minority variants present at various levels in the population. Furthermore, we found that overall diversity was dramatically reduced after single passages in cell lines. Finally, we constructed an infectious clone from this outbreak and identified a novel 3' untranslated region (UTR) structure, not previously found in nature, that led to increased replication in insect cells.
Conclusions/significance: Here we preformed an intrahost quasispecies analysis of the new CHIKV outbreak in the Caribbean. We identified novel variants present in infected individuals, as well as a new 3'UTR structure, suggesting that CHIKV has rapidly evolved in a short period of time once it entered this naïve population. These studies highlight the need to continue viral diversity surveillance over time as this epidemic evolves in order to understand the evolutionary potential of CHIKV.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Van Bortel W, Dorleans F, Rosine J, Blateau A, Rousset D, et al. (2014) Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Euro surveillance: bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin 19. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
