

Btn2a2, a T cell immunomodulatory molecule coregulated with MHC class II genes
- PMID: 26809444
- PMCID: PMC4749920
- DOI: 10.1084/jem.20150435
Btn2a2, a T cell immunomodulatory molecule coregulated with MHC class II genes
Abstract
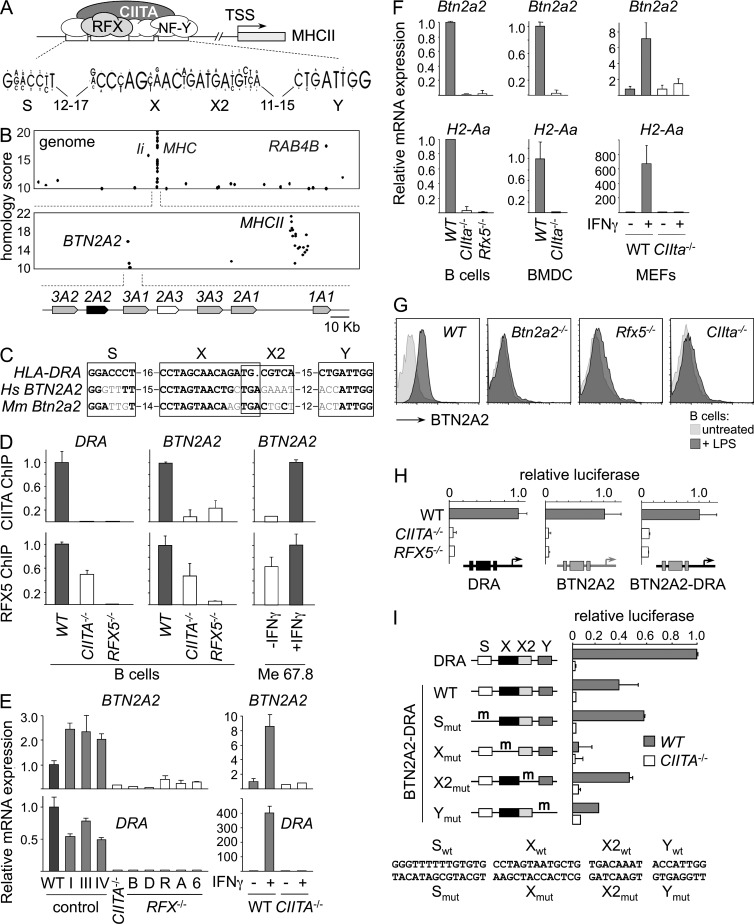
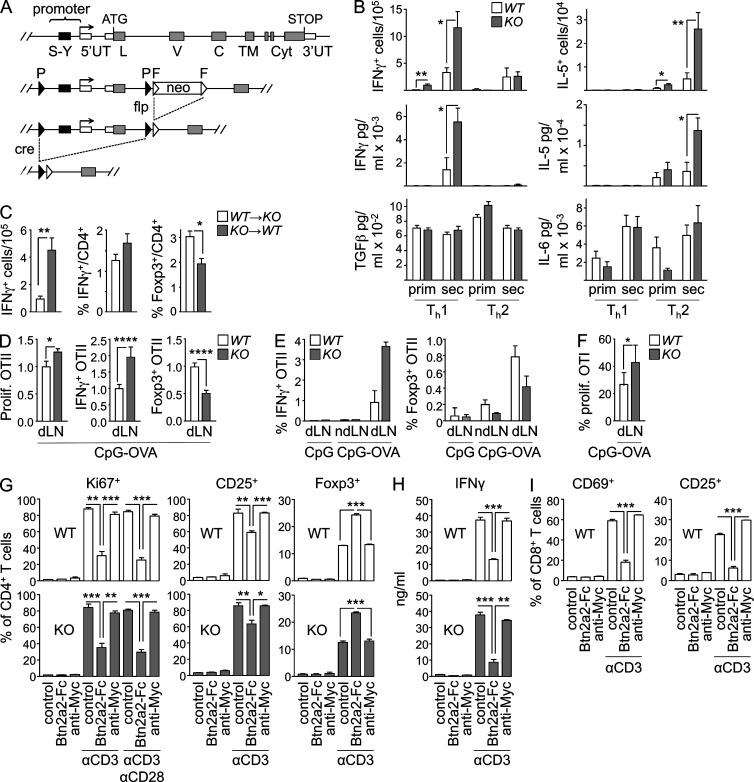
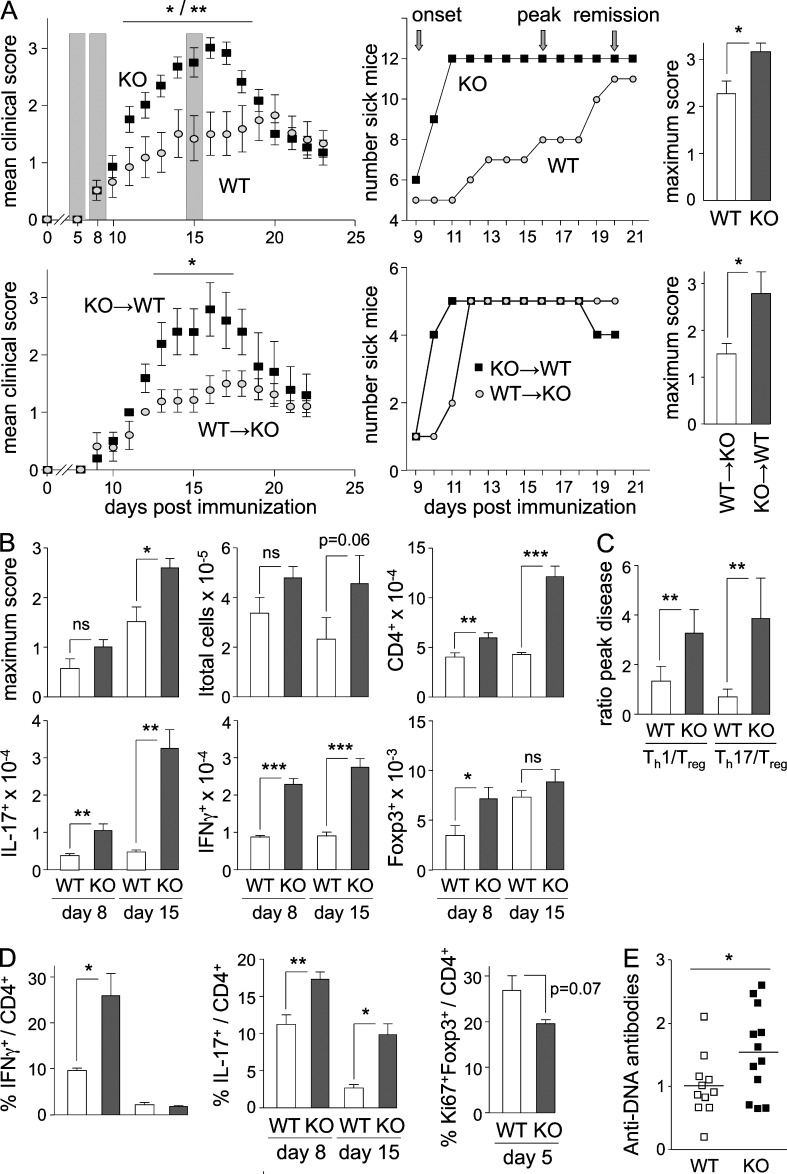
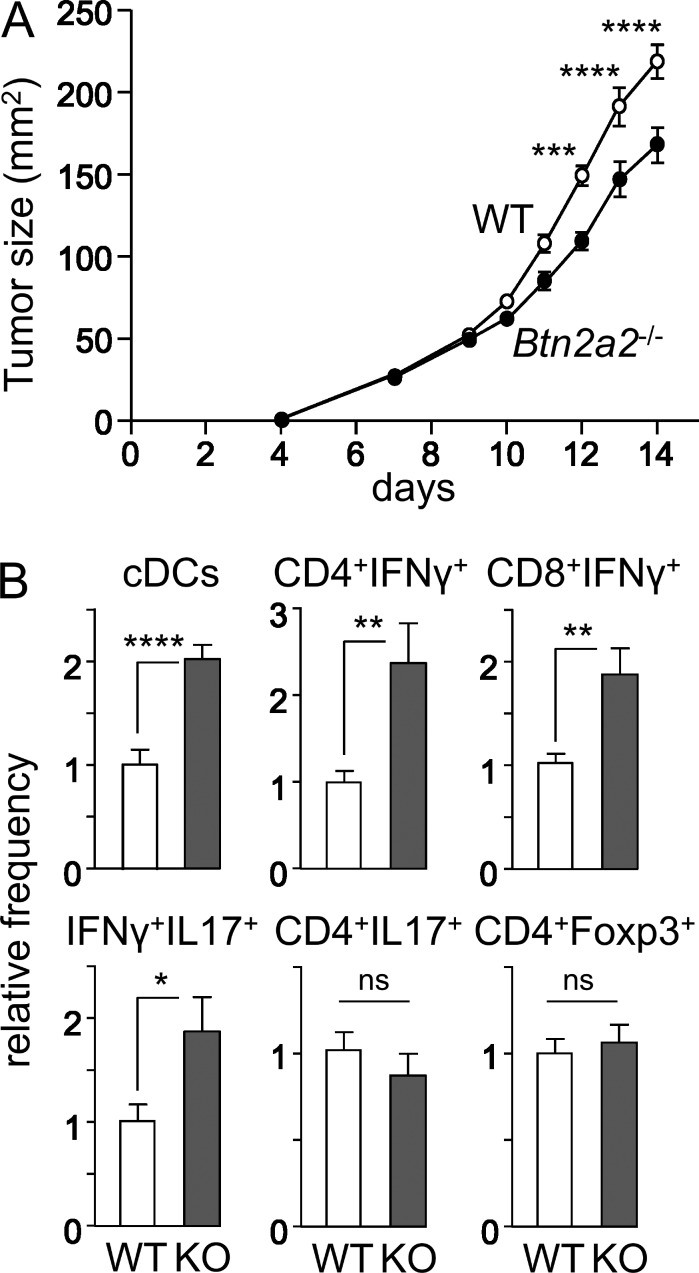
Evidence has recently emerged that butyrophilins, which are members of the extended B7 family of co-stimulatory molecules, have diverse functions in the immune system. We found that the human and mouse genes encoding butyrophilin-2A2 (BTN2A2) are regulated by the class II trans-activator and regulatory factor X, two transcription factors dedicated to major histocompatibility complex class II expression, suggesting a role in T cell immunity. To address this, we generated Btn2a2-deficient mice. Btn2a2(-/-) mice exhibited enhanced effector CD4(+) and CD8(+) T cell responses, impaired CD4(+) regulatory T cell induction, potentiated antitumor responses, and exacerbated experimental autoimmune encephalomyelitis. Altered immune responses were attributed to Btn2a2 deficiency in antigen-presenting cells rather than T cells or nonhematopoietic cells. These results provide the first genetic evidence that BTN2A2 is a co-inhibitory molecule that modulates T cell-mediated immunity.
© 2016 Sarter et al.
Figures
References
-
- Arnett H.A., Escobar S.S., Gonzalez-Suarez E., Budelsky A.L., Steffen L.A., Boiani N., Zhang M., Siu G., Brewer A.W., and Viney J.L.. 2007. BTNL2, a butyrophilin/B7-like molecule, is a negative costimulatory molecule modulated in intestinal inflammation. J. Immunol. 178:1523–1533. 10.4049/jimmunol.178.3.1523 - DOI - PubMed
-
- Azijli K., Stelloo E., Peters G.J., and VAN DEN Eertwegh A.J.. 2014. New developments in the treatment of metastatic melanoma: immune checkpoint inhibitors and targeted therapies. Anticancer Res. 34:1493–1505. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
