

Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer
- PMID: 26813288
- PMCID: PMC4728783
- DOI: 10.1186/s13059-016-0879-2
Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer
Abstract
Background: One of the hallmarks of cancer is the disruption of gene expression patterns. Many molecular lesions contribute to this phenotype, and the importance of aberrant DNA methylation profiles is increasingly recognized. Much of the research effort in this area has examined proximal promoter regions and epigenetic alterations at other loci are not well characterized.
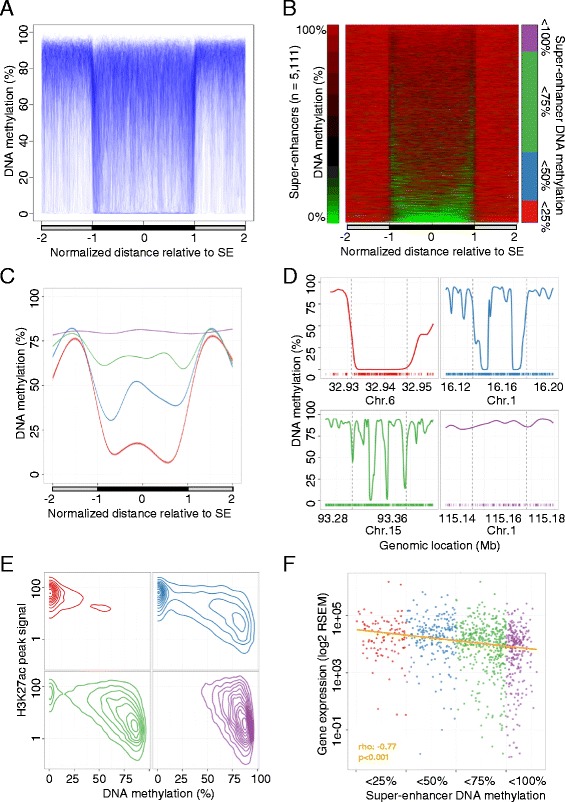
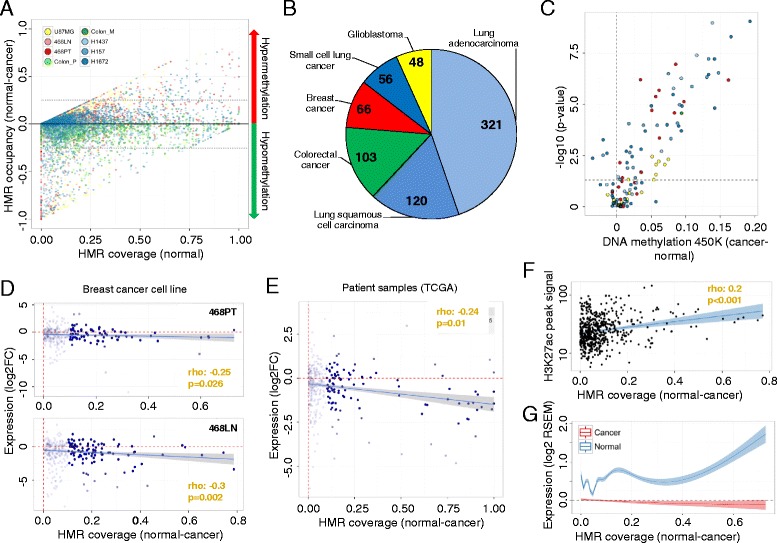
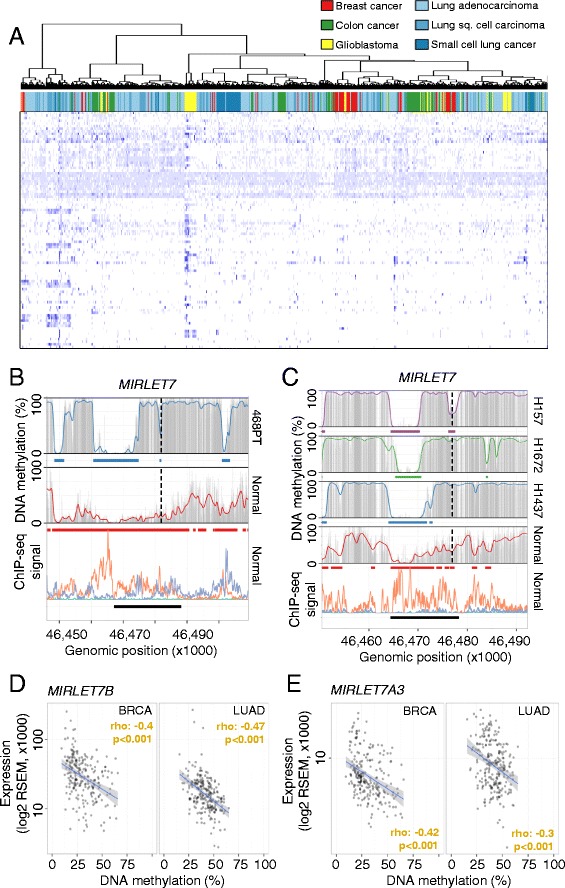
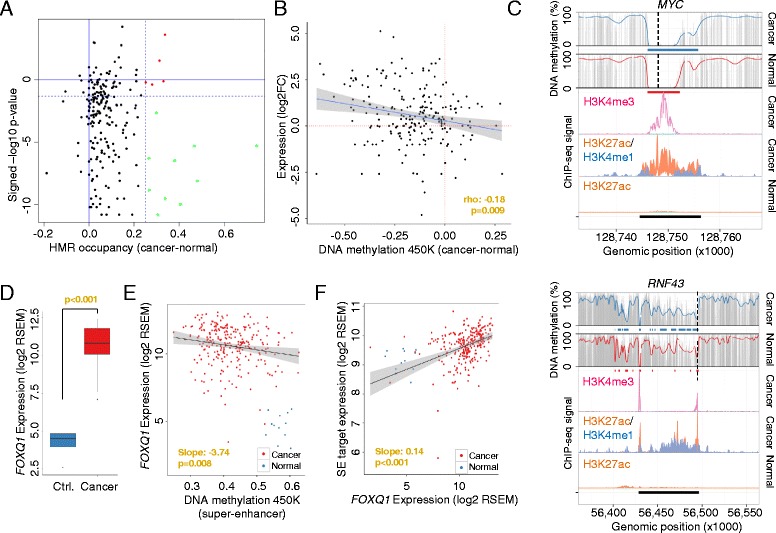
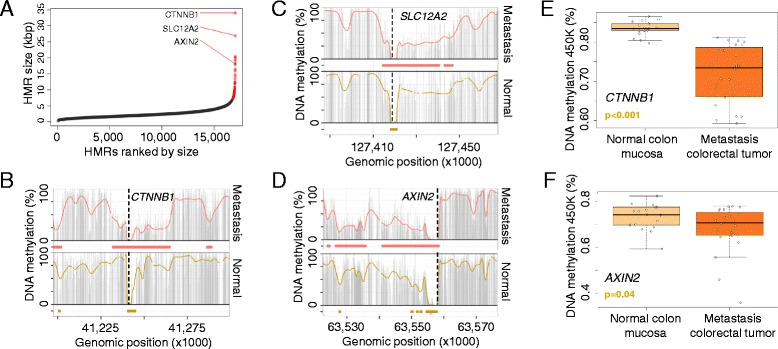
Results: Using whole genome bisulfite sequencing to examine uncharted regions of the epigenome, we identify a type of far-reaching DNA methylation alteration in cancer cells of the distal regulatory sequences described as super-enhancers. Human tumors undergo a shift in super-enhancer DNA methylation profiles that is associated with the transcriptional silencing or the overactivation of the corresponding target genes. Intriguingly, we observe locally active fractions of super-enhancers detectable through hypomethylated regions that suggest spatial variability within the large enhancer clusters. Functionally, the DNA methylomes obtained suggest that transcription factors contribute to this local activity of super-enhancers and that trans-acting factors modulate DNA methylation profiles with impact on transforming processes during carcinogenesis.
Conclusions: We develop an extensive catalogue of human DNA methylomes at base resolution to better understand the regulatory functions of DNA methylation beyond those of proximal promoter gene regions. CpG methylation status in normal cells points to locally active regulatory sites at super-enhancers, which are targeted by specific aberrant DNA methylation events in cancer, with putative effects on the expression of downstream genes.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
