

Review
doi: 10.1186/s13059-016-0881-8.
A survey of best practices for RNA-seq data analysis
Affiliations
- PMID: 26813401
- PMCID: PMC4728800
- DOI: 10.1186/s13059-016-0881-8
Item in Clipboard
Review
A survey of best practices for RNA-seq data analysis
Genome Biol.
.
Erratum in
-
Erratum to: A survey of best practices for RNA-seq data analysis.Genome Biol. 2016 Aug 26;17(1):181. doi: 10.1186/s13059-016-1047-4. Genome Biol. 2016. PMID: 27565134 Free PMC article. No abstract available.
Abstract
RNA-sequencing (RNA-seq) has a wide variety of applications, but no single analysis pipeline can be used in all cases. We review all of the major steps in RNA-seq data analysis, including experimental design, quality control, read alignment, quantification of gene and transcript levels, visualization, differential gene expression, alternative splicing, functional analysis, gene fusion detection and eQTL mapping. We highlight the challenges associated with each step. We discuss the analysis of small RNAs and the integration of RNA-seq with other functional genomics techniques. Finally, we discuss the outlook for novel technologies that are changing the state of the art in transcriptomics.
Figures
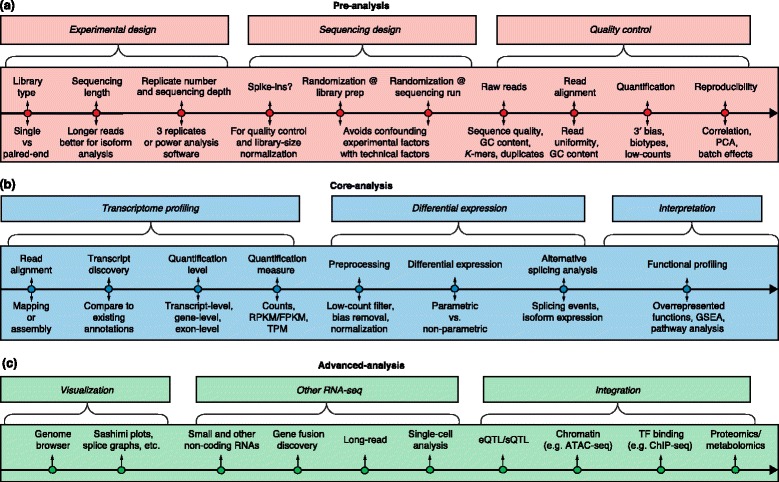
A generic roadmap for RNA-seq computational analyses. The major analysis steps are listed above the lines for pre-analysis, core analysis and advanced analysis. The key analysis issues for each step that are listed below the lines are discussed in the text. a Preprocessing includes experimental design, sequencing design, and quality control steps. b Core analyses include transcriptome profiling, differential gene expression, and functional profiling. c Advanced analysis includes visualization, other RNA-seq technologies, and data integration. Abbreviations: ChIP-seq Chromatin immunoprecipitation sequencing, eQTL Expression quantitative loci, FPKM Fragments per kilobase of exon model per million mapped reads, GSEA Gene set enrichment analysis, PCA Principal component analysis, RPKM Reads per kilobase of exon model per million reads, sQTL Splicing quantitative trait loci, TF Transcription factor, TPM Transcripts per million
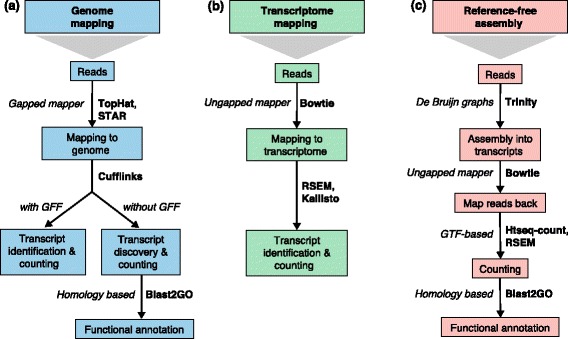
Read mapping and transcript identification strategies. Three basic strategies for regular RNA-seq analysis. a An annotated genome is available and reads are mapped to the genome with a gapped mapper. Next (novel) transcript discovery and quantification can proceed with or without an annotation file. Novel transcripts are then functionally annotated. b If no novel transcript discovery is needed, reads can be mapped to the reference transcriptome using an ungapped aligner. Transcript identification and quantification can occur simultaneously. c When no genome is available, reads need to be assembled first into contigs or transcripts. For quantification, reads are mapped back to the novel reference transcriptome and further analysis proceeds as in (b) followed by the functional annotation of the novel transcripts as in (a). Representative software that can be used at each analysis step are indicated in bold text. Abbreviations: GFF General Feature Format, GTF gene transfer format, RSEM RNA-Seq by Expectation Maximization
