

Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity
- PMID: 26816666
- PMCID: PMC4723237
- DOI: 10.14336/AD.2015.0702
Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity
Abstract
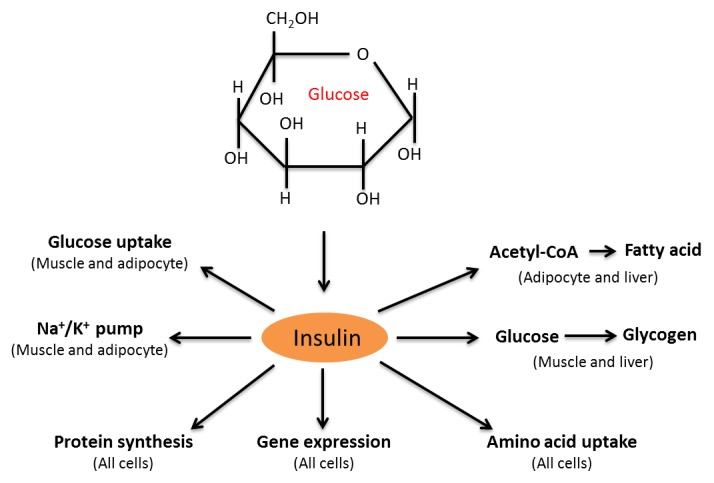
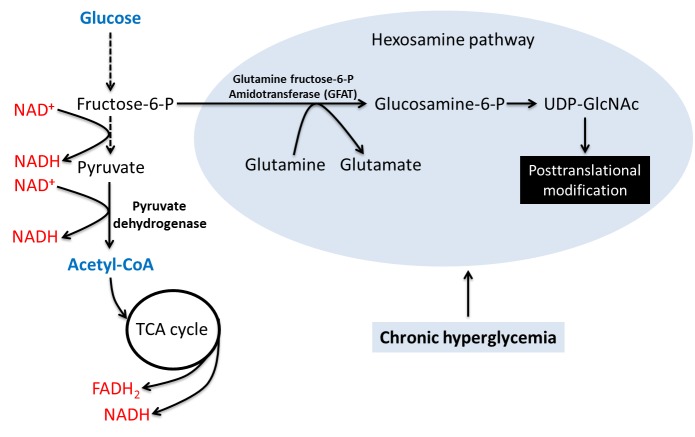
Diabetes and its complications are caused by chronic glucotoxicity driven by persistent hyperglycemia. In this article, we review the mechanisms of diabetic glucotoxicity by focusing mainly on hyperglycemic stress and carbon stress. Mechanisms of hyperglycemic stress include reductive stress or pseudohypoxic stress caused by redox imbalance between NADH and NAD(+) driven by activation of both the polyol pathway and poly ADP ribose polymerase; the hexosamine pathway; the advanced glycation end products pathway; the protein kinase C activation pathway; and the enediol formation pathway. Mechanisms of carbon stress include excess production of acetyl-CoA that can over-acetylate a proteome and excess production of fumarate that can over-succinate a proteome; both of which can increase glucotoxicity in diabetes. For hyperglycemia stress, we also discuss the possible role of mitochondrial complex I in diabetes as this complex, in charge of NAD(+) regeneration, can make more reactive oxygen species (ROS) in the presence of excess NADH. For carbon stress, we also discuss the role of sirtuins in diabetes as they are deacetylases that can reverse protein acetylation thereby attenuating diabetic glucotoxicity and improving glucose metabolism. It is our belief that targeting some of the stress pathways discussed in this article may provide new therapeutic strategies for treatment of diabetes and its complications.
Keywords: carbon stress; diabetes; glucotoxicity; hyperglycemic stress; pseudohypoxia; reactive oxygen species; redox imbalance.
Figures






Similar articles
-
Role of pseudohypoxia in the pathogenesis of type 2 diabetes.Hypoxia (Auckl). 2019 Jun 5;7:33-40. doi: 10.2147/HP.S202775. eCollection 2019. Hypoxia (Auckl). 2019. PMID: 31240235 Free PMC article.
-
Protein Modifications as Manifestations of Hyperglycemic Glucotoxicity in Diabetes and Its Complications.Biochem Insights. 2016 Mar 23;9:1-9. doi: 10.4137/BCI.S36141. eCollection 2016. Biochem Insights. 2016. PMID: 27042090 Free PMC article. Review.
-
Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications.Diabetes Metab Syndr Obes. 2016 May 10;9:145-53. doi: 10.2147/DMSO.S106087. eCollection 2016. Diabetes Metab Syndr Obes. 2016. PMID: 27274295 Free PMC article. Review.
-
Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders.Eur J Nutr. 2016 Dec;55(8):2339-2345. doi: 10.1007/s00394-016-1212-2. Epub 2016 Apr 15. Eur J Nutr. 2016. PMID: 27084094 Free PMC article. Review.
-
The Role of Oxidative Stress in Diabetic Neuropathy: Generation of Free Radical Species in the Glycation Reaction and Gene Polymorphisms Encoding Antioxidant Enzymes to Genetic Susceptibility to Diabetic Neuropathy in Population of Type I Diabetic Patients.Cell Biochem Biophys. 2015 Apr;71(3):1425-43. doi: 10.1007/s12013-014-0365-y. Cell Biochem Biophys. 2015. PMID: 25427889
Cited by
-
SIRT6's function in controlling the metabolism of lipids and glucose in diabetic nephropathy.Front Endocrinol (Lausanne). 2023 Oct 9;14:1244705. doi: 10.3389/fendo.2023.1244705. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 37876546 Free PMC article. Review.
-
Coadjuvants in the Diabetic Complications: Nutraceuticals and Drugs with Pleiotropic Effects.Int J Mol Sci. 2016 Aug 5;17(8):1273. doi: 10.3390/ijms17081273. Int J Mol Sci. 2016. PMID: 27527163 Free PMC article. Review.
-
Metabolic Responses to Reductive Stress.Antioxid Redox Signal. 2020 Jun;32(18):1330-1347. doi: 10.1089/ars.2019.7803. Epub 2019 Jul 18. Antioxid Redox Signal. 2020. PMID: 31218894 Free PMC article. Review.
-
Reexploring 5-methoxyindole-2-carboxylic acid (MICA) as a potential antidiabetic agent.Diabetes Metab Syndr Obes. 2018 May 4;11:183-186. doi: 10.2147/DMSO.S166485. eCollection 2018. Diabetes Metab Syndr Obes. 2018. PMID: 29765243 Free PMC article.
-
Novel drugs affecting diabetic peripheral neuropathy.Iran J Basic Med Sci. 2024;27(6):657-670. doi: 10.22038/IJBMS.2024.75367.16334. Iran J Basic Med Sci. 2024. PMID: 38645500 Free PMC article. Review.
References
-
- Bensellam M,Laybutt DR,Jonas JC (2012). The molecular mechanisms of pancreatic beta-cell glucotoxicity: recent findings and future research directions. Molecular and cellular endocrinology, 364: 1-27 - PubMed
-
- Del Prato S (2009). Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet Med, 26: 1185-1192 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources