

Predicting transcription factor site occupancy using DNA sequence intrinsic and cell-type specific chromatin features
- PMID: 26818008
- PMCID: PMC4895346
- DOI: 10.1186/s12859-015-0846-z
Predicting transcription factor site occupancy using DNA sequence intrinsic and cell-type specific chromatin features
Abstract
Background: Understanding the mechanisms by which transcription factors (TF) are recruited to their physiological target sites is crucial for understanding gene regulation. DNA sequence intrinsic features such as predicted binding affinity are often not very effective in predicting in vivo site occupancy and in any case could not explain cell-type specific binding events. Recent reports show that chromatin accessibility, nucleosome occupancy and specific histone post-translational modifications greatly influence TF site occupancy in vivo. In this work, we use machine-learning methods to build predictive models and assess the relative importance of different sequence-intrinsic and chromatin features in the TF-to-target-site recruitment process.
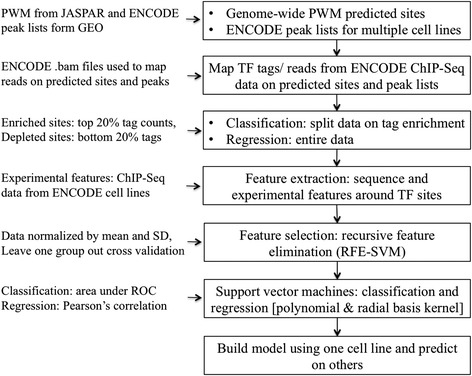
Methods: Our study primarily relies on recent data published by the ENCODE consortium. Five dissimilar TFs assayed in multiple cell-types were selected as examples: CTCF, JunD, REST, GABP and USF2. We used two types of candidate target sites: (a) predicted sites obtained by scanning the whole genome with a position weight matrix, and (b) cell-type specific peak lists provided by ENCODE. Quantitative in vivo occupancy levels in different cell-types were based on ChIP-seq data for the corresponding TFs. In parallel, we computed a number of associated sequence-intrinsic and experimental features (histone modification, DNase I hypersensitivity, etc.) for each site. Machine learning algorithms were then used in a binary classification and regression framework to predict site occupancy and binding strength, for the purpose of assessing the relative importance of different contextual features.
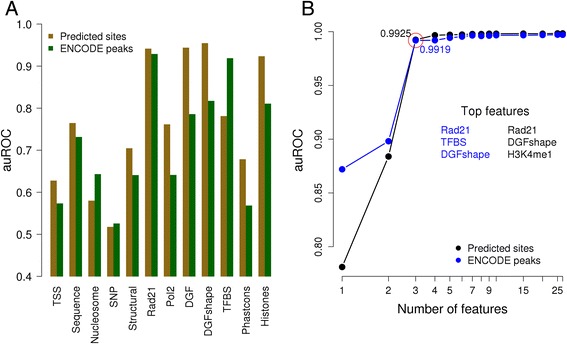
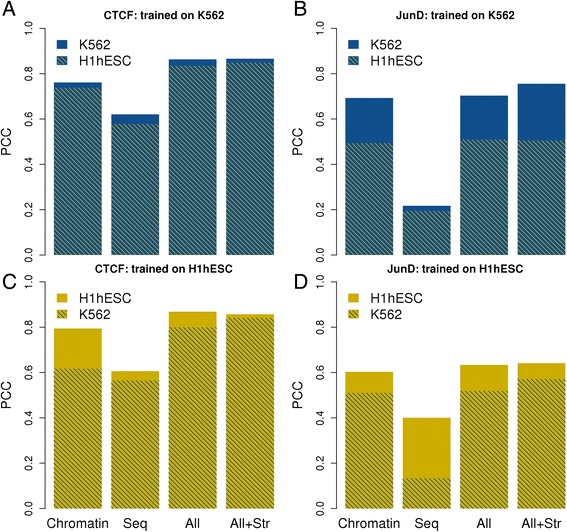
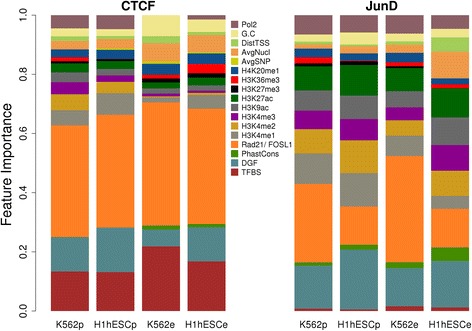
Results: We observed striking differences in the feature importance rankings between the five factors tested. PWM-scores were amongst the most important features only for CTCF and REST but of little value for JunD and USF2. Chromatin accessibility and active histone marks are potent predictors for all factors except REST. Structural DNA parameters, repressive and gene body associated histone marks are generally of little or no predictive value.
Conclusions: We define a general and extensible computational framework for analyzing the importance of various DNA-intrinsic and chromatin-associated features in determining cell-type specific TF binding to target sites. The application of our methodology to ENCODE data has led to new insights on transcription regulatory processes and may serve as example for future studies encompassing even larger datasets.
Figures
Similar articles
-
Assessing the model transferability for prediction of transcription factor binding sites based on chromatin accessibility.BMC Bioinformatics. 2017 Jul 27;18(1):355. doi: 10.1186/s12859-017-1769-7. BMC Bioinformatics. 2017. PMID: 28750606 Free PMC article.
-
Modeling co-occupancy of transcription factors using chromatin features.Nucleic Acids Res. 2016 Mar 18;44(5):e49. doi: 10.1093/nar/gkv1281. Epub 2015 Nov 20. Nucleic Acids Res. 2016. PMID: 26590261 Free PMC article.
-
BinDNase: a discriminatory approach for transcription factor binding prediction using DNase I hypersensitivity data.Bioinformatics. 2015 Sep 1;31(17):2852-9. doi: 10.1093/bioinformatics/btv294. Epub 2015 May 7. Bioinformatics. 2015. PMID: 25957350
-
Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond.Cell Cycle. 2014;13(18):2847-52. doi: 10.4161/15384101.2014.949201. Cell Cycle. 2014. PMID: 25486472 Free PMC article. Review.
-
Sequence and chromatin determinants of transcription factor binding and the establishment of cell type-specific binding patterns.Biochim Biophys Acta Gene Regul Mech. 2020 Jun;1863(6):194443. doi: 10.1016/j.bbagrm.2019.194443. Epub 2019 Oct 19. Biochim Biophys Acta Gene Regul Mech. 2020. PMID: 31639474 Free PMC article. Review.
Cited by
-
SemanticCAP: Chromatin Accessibility Prediction Enhanced by Features Learning from a Language Model.Genes (Basel). 2022 Mar 23;13(4):568. doi: 10.3390/genes13040568. Genes (Basel). 2022. PMID: 35456374 Free PMC article.
-
An interpretable bimodal neural network characterizes the sequence and preexisting chromatin predictors of induced transcription factor binding.Genome Biol. 2021 Jan 7;22(1):20. doi: 10.1186/s13059-020-02218-6. Genome Biol. 2021. PMID: 33413545 Free PMC article.
-
Cross-Cell-Type Prediction of TF-Binding Site by Integrating Convolutional Neural Network and Adversarial Network.Int J Mol Sci. 2019 Jul 12;20(14):3425. doi: 10.3390/ijms20143425. Int J Mol Sci. 2019. PMID: 31336830 Free PMC article.
-
MTTFsite: cross-cell type TF binding site prediction by using multi-task learning.Bioinformatics. 2019 Dec 15;35(24):5067-5077. doi: 10.1093/bioinformatics/btz451. Bioinformatics. 2019. PMID: 31161194 Free PMC article.
-
Assessing the model transferability for prediction of transcription factor binding sites based on chromatin accessibility.BMC Bioinformatics. 2017 Jul 27;18(1):355. doi: 10.1186/s12859-017-1769-7. BMC Bioinformatics. 2017. PMID: 28750606 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
