

Potential Therapeutic Benefits of Maintaining Mitochondrial Health in Peripheral Neuropathies
- PMID: 26818748
- PMCID: PMC4981743
- DOI: 10.2174/1570159x14666151126215358
Potential Therapeutic Benefits of Maintaining Mitochondrial Health in Peripheral Neuropathies
Abstract
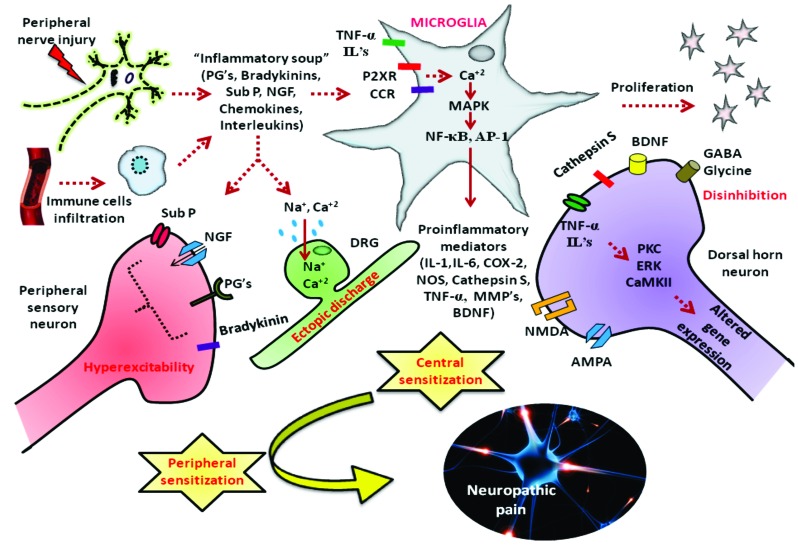
Background: Peripheral neuropathies are a group of diseases characterized by malfunctioning of peripheral nervous system. Neuropathic pain, one of the core manifestations of peripheral neuropathy remains as the most severe disabling condition affecting the social and daily routine life of patients suffering from peripheral neuropathy.
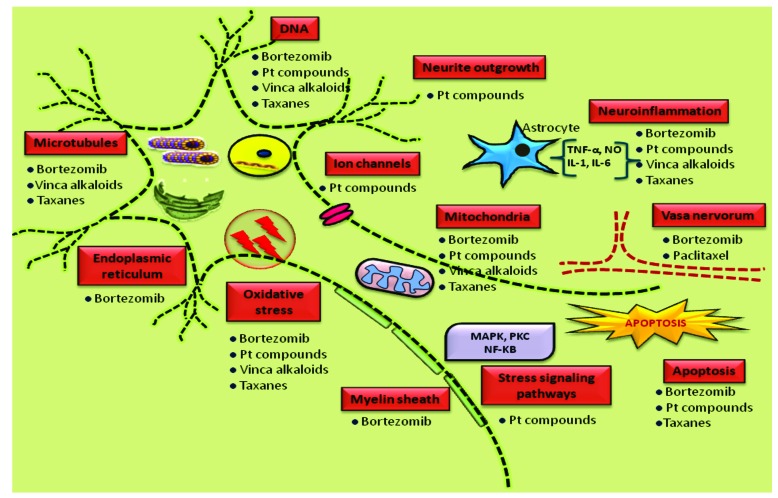
Method: The current review is aimed at unfolding the possible role of mitochondrial dysfunction in peripheral nerve damage and to discuss on the probable therapeutic strategies against neuronal mitotoxicity. The article also highlights the therapeutic significance of maintaining a healthy mitochondrial environment in neuronal cells via pharmacological management in context of peripheral neuropathies.
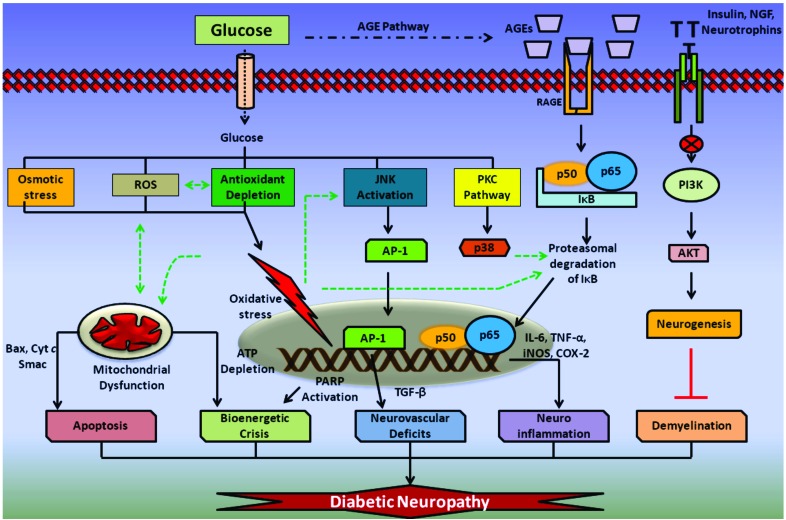
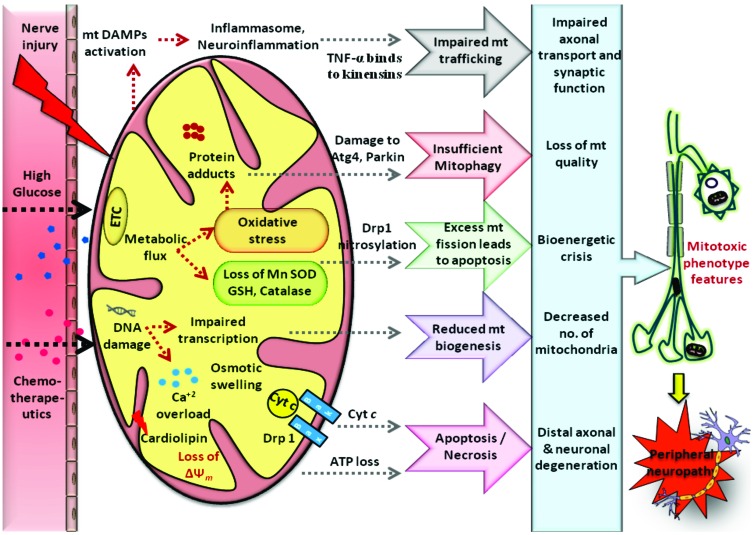
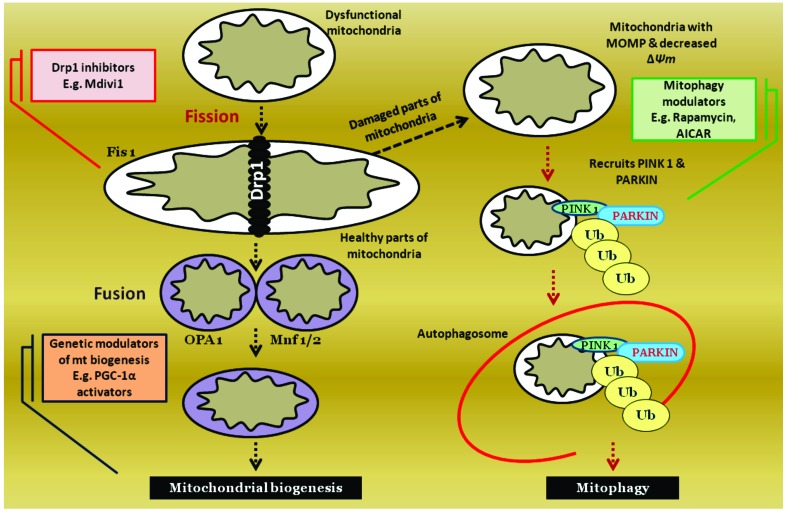
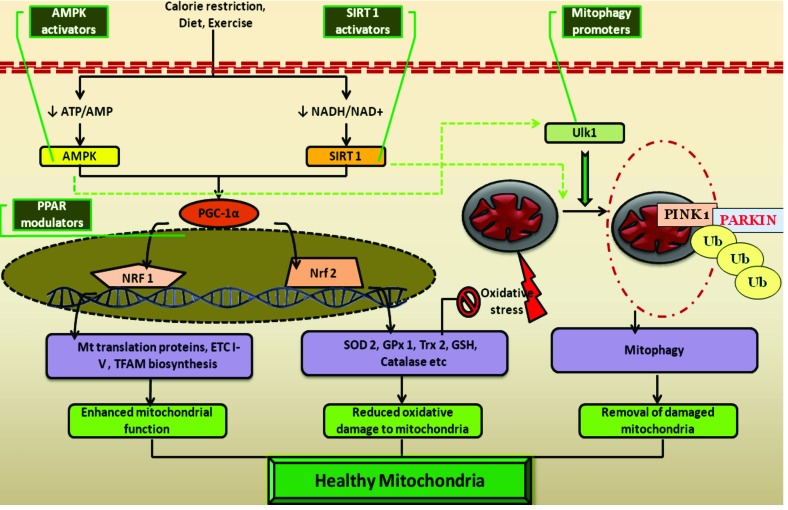
Results: Aberrant cellular signaling coupled with changes in neurotransmission, peripheral and central sensitization are found to be responsible for the pathogenesis of variant toxic neuropathies. Current research reports have indicated the possible involvement of mitochondria mediated redox imbalance as one of the principal causes of neuropathy aetiologies. In addition to imbalance in redox homeostasis, mitochondrial dysfunction is also responsible for alterations in physiological bioenergetic metabolism, apoptosis and autophagy pathways.
Conclusions: In spite of various etiological factors, mitochondrial dysfunction has been found to be a major pathomechanism underlying the neuronal dysfunction associated with peripheral neuropathies. Pharmacological modulation of mitochondria either directly or indirectly is expected to yield therapeutic relief from various primary and secondary mitochondrial diseases.
Figures
References
-
- Ganesh Yerra V., Negi G., Sharma S.S., Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013;1:394–397. [http://dx.doi.org/10.1016/j.redox.2013.07.005]. [PMID: 24024177]. - PMC - PubMed
-
- Baron R., Binder A., Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9(8):807–819. [http://dx.doi.org/10.1016/S1474-4422(10) 70143-5]. [PMID: 20650402]. - PubMed
-
- Weisberg L.A., Garcia C., Strub R.L. Essentials of clinical neurology. St Louis: Mosby; 1996. Diseases of the peripheral nerves and motor neurons. pp. 458–494.
-
- Saporta M.A., Shy M.E. Inherited peripheral neuropathies. Neurol. Clin. 2013;31(2):597–619. [http://dx.doi.org/10.1016/ j.ncl.2013.01.009]. [PMID: 23642725]. - PMC - PubMed
-
- Carelli V., Ross-Cisneros F.N., Sadun A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004;23(1):53–89. [http://dx.doi.org/10.1016/j.preteyeres.2003.10. 003]. [PMID: 14766317]. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical