

SOHSite: incorporating evolutionary information and physicochemical properties to identify protein S-sulfenylation sites
- PMID: 26819243
- PMCID: PMC4895302
- DOI: 10.1186/s12864-015-2299-1
SOHSite: incorporating evolutionary information and physicochemical properties to identify protein S-sulfenylation sites
Abstract
Background: Protein S-sulfenylation is a type of post-translational modification (PTM) involving the covalent binding of a hydroxyl group to the thiol of a cysteine amino acid. Recent evidence has shown the importance of S-sulfenylation in various biological processes, including transcriptional regulation, apoptosis and cytokine signaling. Determining the specific sites of S-sulfenylation is fundamental to understanding the structures and functions of S-sulfenylated proteins. However, the current lack of reliable tools often limits researchers to use expensive and time-consuming laboratory techniques for the identification of S-sulfenylation sites. Thus, we were motivated to develop a bioinformatics method for investigating S-sulfenylation sites based on amino acid compositions and physicochemical properties.
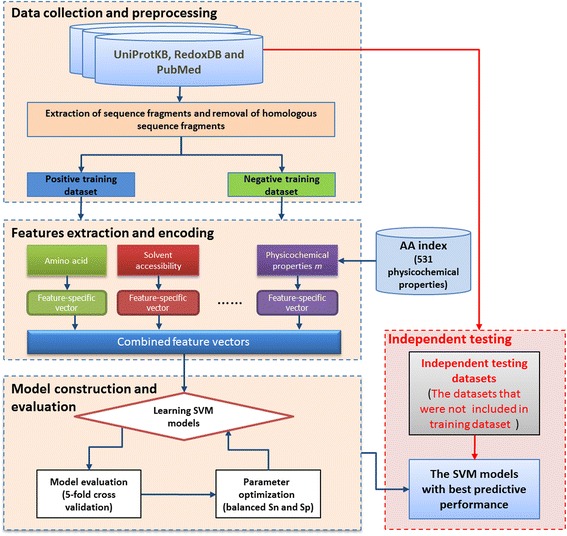
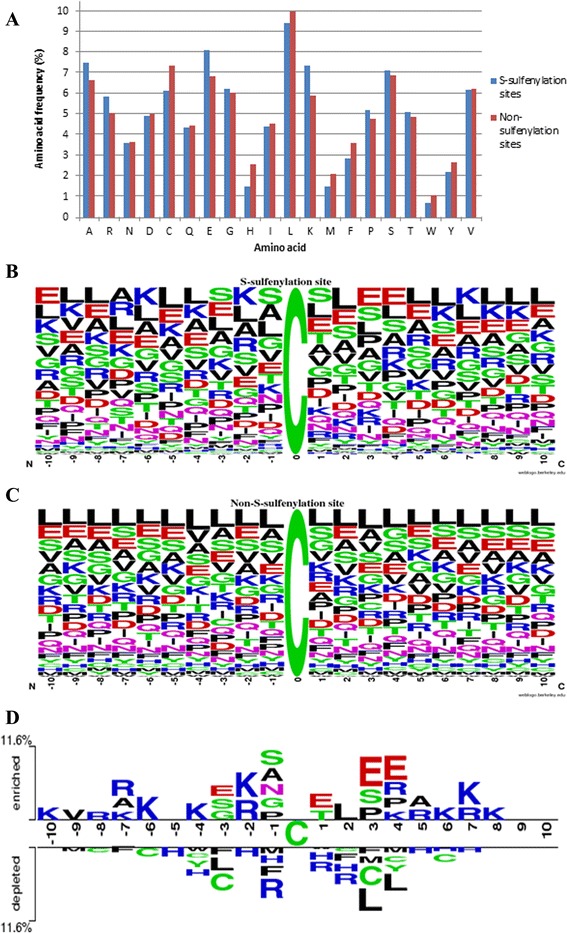
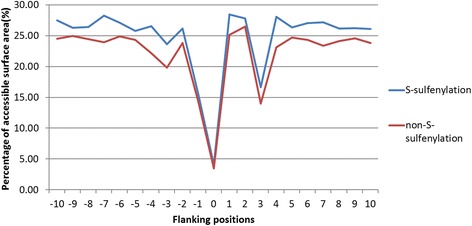
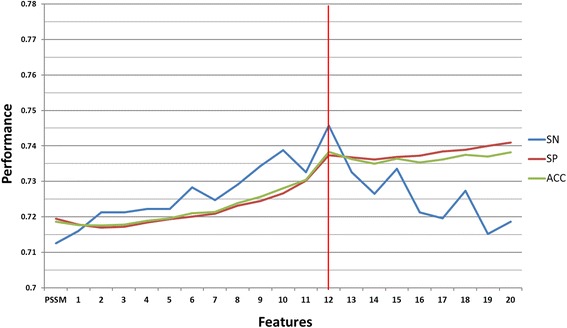
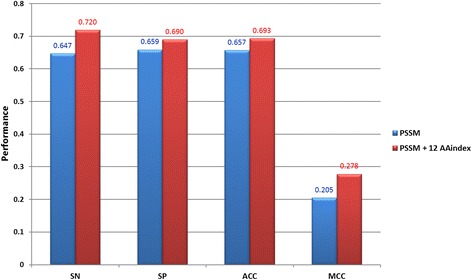
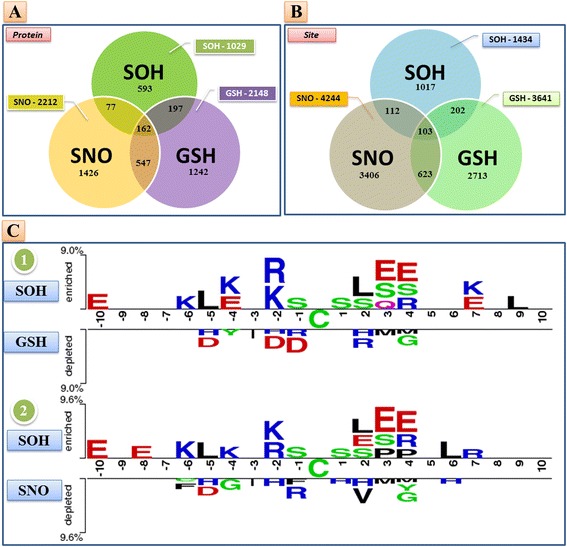
Results: In this work, physicochemical properties were utilized not only to identify S-sulfenylation sites from 1,096 experimentally verified S-sulfenylated proteins, but also to compare the effectiveness of prediction with other characteristics such as amino acid composition (AAC), amino acid pair composition (AAPC), solvent-accessible surface area (ASA), amino acid substitution matrix (BLOSUM62), position-specific scoring matrix (PSSM), and positional weighted matrix (PWM). Various prediction models were built using support vector machine (SVM) and evaluated by five-fold cross-validation. The model constructed from hybrid features, including PSSM and physicochemical properties, yielded the best performance with sensitivity, specificity, accuracy and MCC measurements of 0.746, 0.737, 0.738 and 0.337, respectively. The selected model also provided a promising accuracy (0.693) on an independent testing dataset. Additionally, we employed TwoSampleLogo to help discover the difference of amino acid composition among S-sulfenylation, S-glutathionylation and S-nitrosylation sites.
Conclusion: This work proposed a computational method to explore informative features and functions for protein S-sulfenylation. Evaluation by five-fold cross validation indicated that the selected features were effective in the identification of S-sulfenylation sites. Moreover, the independent testing results demonstrated that the proposed method could provide a feasible means for conducting preliminary analyses of protein S-sulfenylation. We also anticipate that the uncovered differences in amino acid composition may facilitate future studies of the extensive crosstalk among S-sulfenylation, S-glutathionylation and S-nitrosylation.
Figures
Similar articles
-
Investigation and identification of protein carbonylation sites based on position-specific amino acid composition and physicochemical features.BMC Bioinformatics. 2017 Mar 14;18(Suppl 3):66. doi: 10.1186/s12859-017-1472-8. BMC Bioinformatics. 2017. PMID: 28361707 Free PMC article.
-
MDD-SOH: exploiting maximal dependence decomposition to identify S-sulfenylation sites with substrate motifs.Bioinformatics. 2016 Jan 15;32(2):165-72. doi: 10.1093/bioinformatics/btv558. Epub 2015 Sep 26. Bioinformatics. 2016. PMID: 26411868
-
Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information.Mol Biosyst. 2017 Nov 21;13(12):2545-2550. doi: 10.1039/c7mb00491e. Mol Biosyst. 2017. PMID: 28990628
-
A Comprehensive Review of In silico Analysis for Protein S-sulfenylation Sites.Protein Pept Lett. 2018;25(9):815-821. doi: 10.2174/0929866525666180905110619. Protein Pept Lett. 2018. PMID: 30182830 Review.
-
Proteome-Wide Analysis of Cysteine S-Sulfenylation Using a Benzothiazine-Based Probe.Curr Protoc Protein Sci. 2019 Feb;95(1):e76. doi: 10.1002/cpps.76. Epub 2018 Oct 12. Curr Protoc Protein Sci. 2019. PMID: 30312022 Free PMC article. Review.
Cited by
-
Characterization and identification of lysine glutarylation based on intrinsic interdependence between positions in the substrate sites.BMC Bioinformatics. 2019 Feb 4;19(Suppl 13):384. doi: 10.1186/s12859-018-2394-9. BMC Bioinformatics. 2019. PMID: 30717647 Free PMC article.
-
MDD-carb: a combinatorial model for the identification of protein carbonylation sites with substrate motifs.BMC Syst Biol. 2017 Dec 21;11(Suppl 7):137. doi: 10.1186/s12918-017-0511-4. BMC Syst Biol. 2017. PMID: 29322938 Free PMC article.
-
dbAMP 2.0: updated resource for antimicrobial peptides with an enhanced scanning method for genomic and proteomic data.Nucleic Acids Res. 2022 Jan 7;50(D1):D460-D470. doi: 10.1093/nar/gkab1080. Nucleic Acids Res. 2022. PMID: 34850155 Free PMC article.
-
DeepCSO: A Deep-Learning Network Approach to Predicting Cysteine S-Sulphenylation Sites.Front Cell Dev Biol. 2020 Dec 1;8:594587. doi: 10.3389/fcell.2020.594587. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33335901 Free PMC article.
-
StackTHPred: Identifying Tumor-Homing Peptides through GBDT-Based Feature Selection with Stacking Ensemble Architecture.Int J Mol Sci. 2023 Jun 19;24(12):10348. doi: 10.3390/ijms241210348. Int J Mol Sci. 2023. PMID: 37373494 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
