

Inhibition of Prostaglandin Reductase 2, a Putative Oncogene Overexpressed in Human Pancreatic Adenocarcinoma, Induces Oxidative Stress-Mediated Cell Death Involving xCT and CTH Gene Expressions through 15-Keto-PGE2
- PMID: 26820738
- PMCID: PMC4731085
- DOI: 10.1371/journal.pone.0147390
Inhibition of Prostaglandin Reductase 2, a Putative Oncogene Overexpressed in Human Pancreatic Adenocarcinoma, Induces Oxidative Stress-Mediated Cell Death Involving xCT and CTH Gene Expressions through 15-Keto-PGE2
Abstract
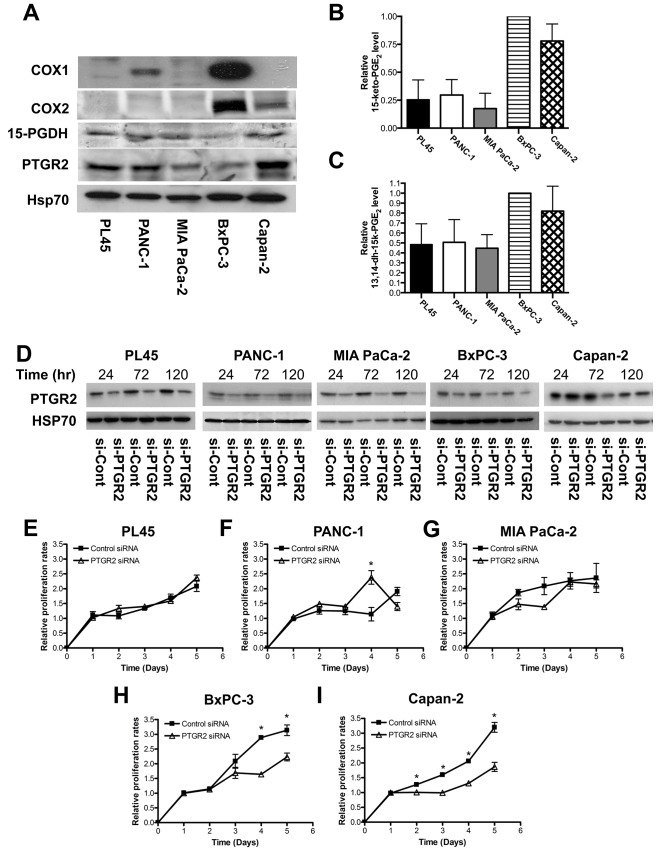
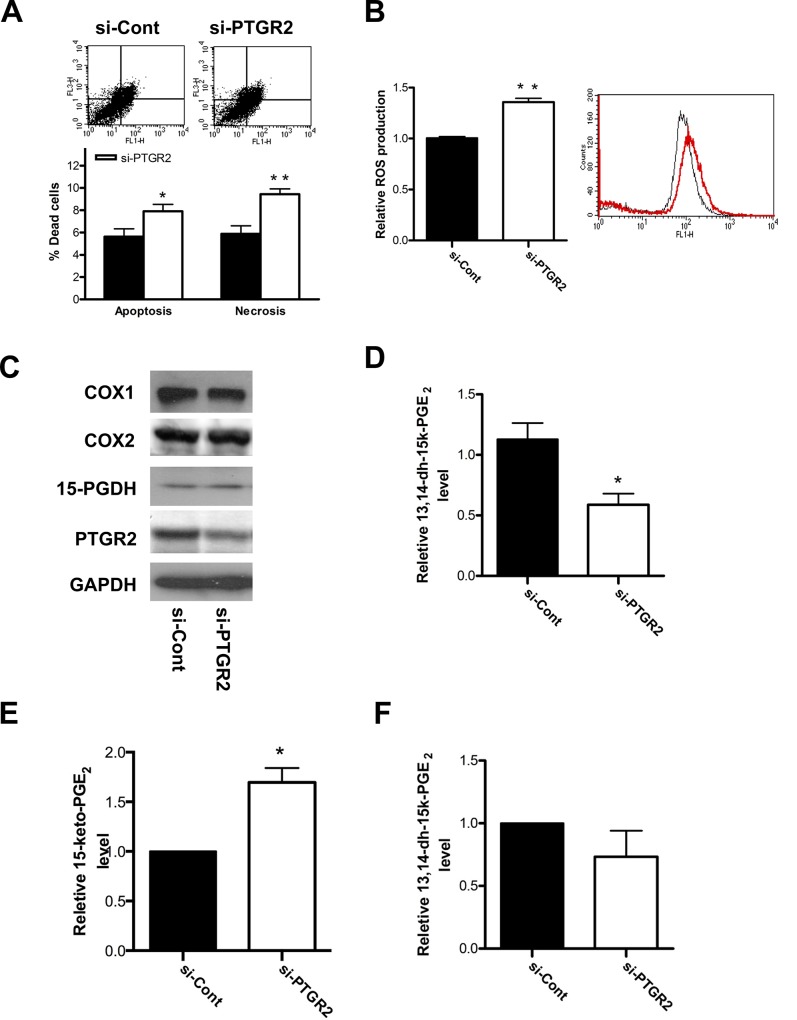
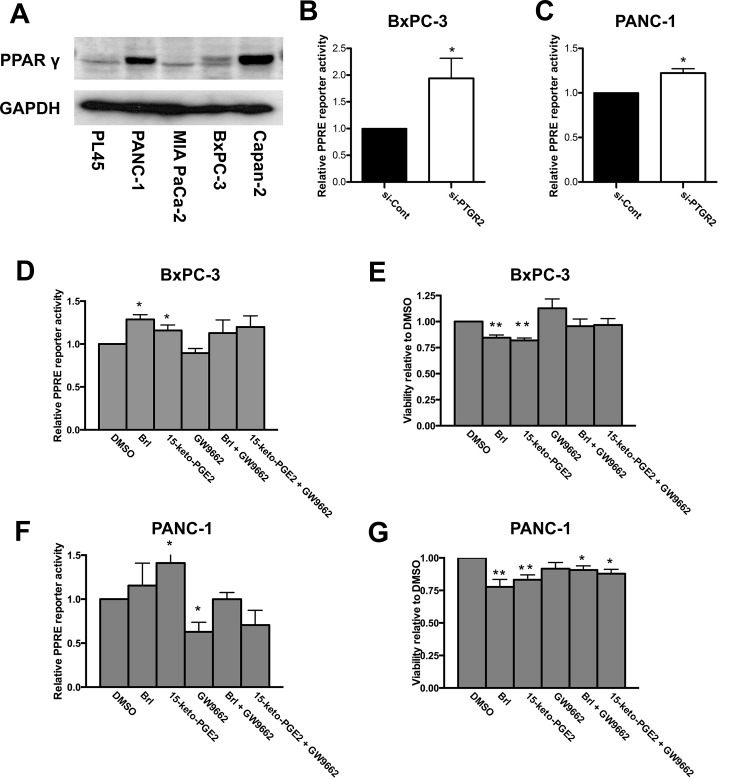
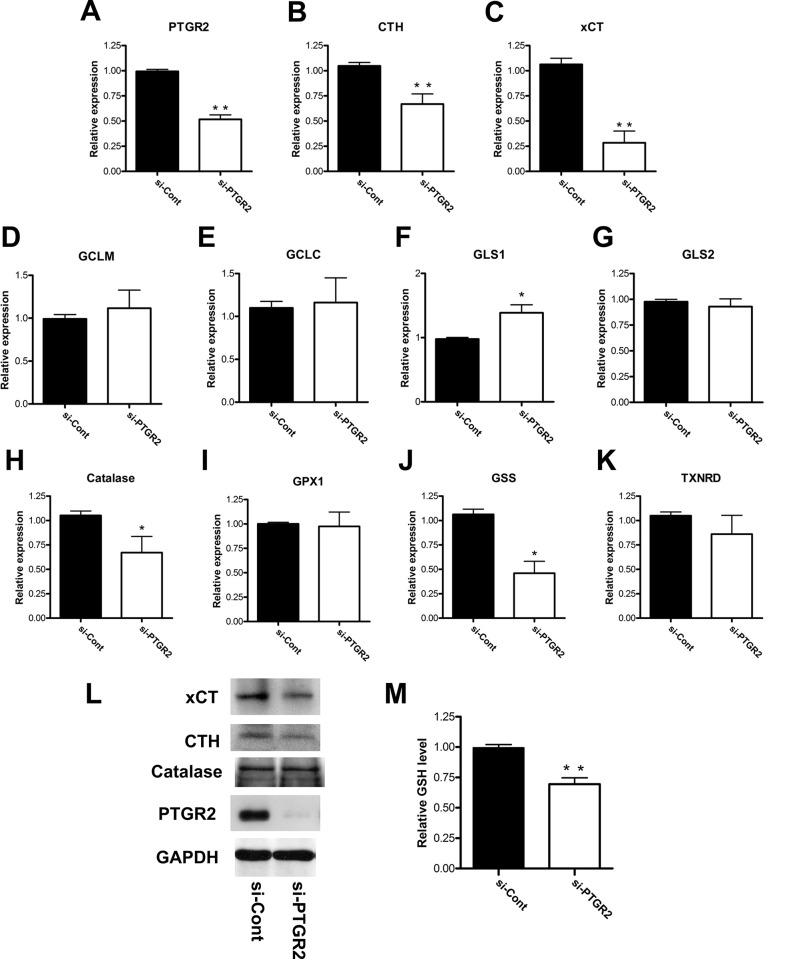
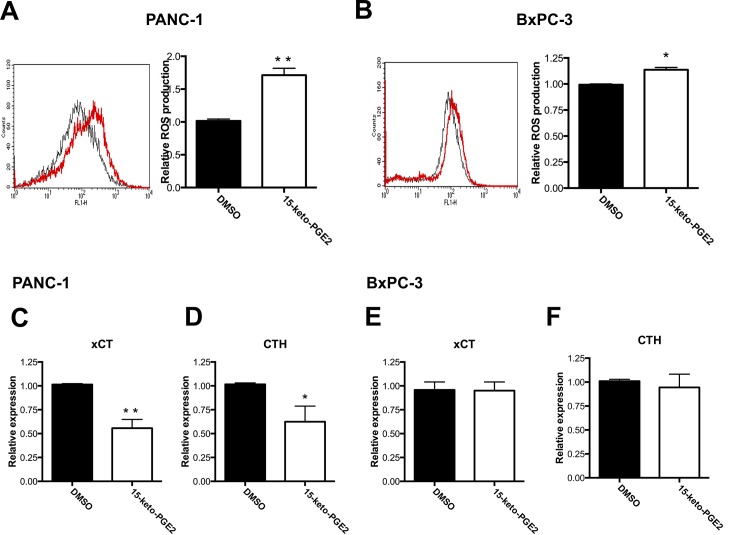
Prostaglandin reductase 2 (PTGR2) is the enzyme that catalyzes 15-keto-PGE2, an endogenous PPARγ ligand, into 13,14-dihydro-15-keto-PGE2. Previously, we have reported a novel oncogenic role of PTGR2 in gastric cancer, where PTGR2 was discovered to modulate ROS-mediated cell death and tumor transformation. In the present study, we demonstrated the oncogenic potency of PTGR2 in pancreatic cancer. First, we observed that the majority of the human pancreatic ductal adenocarcinoma tissues was stained positive for PTGR2 expression but not in the adjacent normal parts. In vitro analyses showed that silencing of PTGR2 expression enhanced ROS production, suppressed pancreatic cell proliferation, and promoted cell death through increasing 15-keto-PGE2. Mechanistically, silencing of PTGR2 or addition of 15-keto-PGE2 suppressed the expressions of solute carrier family 7 member 11 (xCT) and cystathionine gamma-lyase (CTH), two important providers of intracellular cysteine for the generation of glutathione (GSH), which is widely accepted as the first-line antioxidative defense. The oxidative stress-mediated cell death after silencing of PTGR2 or addition of 15-keto-PGE2 was further abolished after restoring intracellular GSH concentrations and cysteine supply by N-acetyl-L-cysteine and 2-Mercaptomethanol. Our data highlight the therapeutic potential of targeting PTGR2/15-keto-PGE2 for pancreatic cancer.
Conflict of interest statement
Figures


Similar articles
-
Prostaglandin reductase 2 modulates ROS-mediated cell death and tumor transformation of gastric cancer cells and is associated with higher mortality in gastric cancer patients.Am J Pathol. 2012 Oct;181(4):1316-26. doi: 10.1016/j.ajpath.2012.07.006. Am J Pathol. 2012. PMID: 22998775
-
Targeting the 15-keto-PGE2-PTGR2 axis modulates systemic inflammation and survival in experimental sepsis.Free Radic Biol Med. 2018 Feb 1;115:113-126. doi: 10.1016/j.freeradbiomed.2017.11.016. Epub 2017 Nov 22. Free Radic Biol Med. 2018. PMID: 29175486
-
Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function.Oncol Rep. 2015 Mar;33(3):1465-74. doi: 10.3892/or.2015.3712. Epub 2015 Jan 13. Oncol Rep. 2015. PMID: 25585997
-
Identification of PTGR2 inhibitors as a new therapeutic strategy for diabetes and obesity.EMBO Mol Med. 2025 May;17(5):938-966. doi: 10.1038/s44321-025-00216-4. Epub 2025 Mar 21. EMBO Mol Med. 2025. PMID: 40119175 Free PMC article.
-
[Cystathionine γ-lyase].Postepy Hig Med Dosw (Online). 2014 Jan 15;68:1-9. doi: 10.5604/17322693.1085372. Postepy Hig Med Dosw (Online). 2014. PMID: 24491890 Review. Polish.
Cited by
-
Discovery of a Cell-Active SuTEx Ligand of Prostaglandin Reductase 2.Chembiochem. 2021 Jun 15;22(12):2134-2139. doi: 10.1002/cbic.202000879. Epub 2021 Apr 29. Chembiochem. 2021. PMID: 33861519 Free PMC article.
-
High-accuracy prediction of colorectal cancer chemotherapy efficacy using machine learning applied to gene expression data.Front Physiol. 2024 Jan 18;14:1272206. doi: 10.3389/fphys.2023.1272206. eCollection 2023. Front Physiol. 2024. PMID: 38304289 Free PMC article.
-
Emerging roles of the solute carrier family in pancreatic cancer.Clin Transl Med. 2021 Mar;11(3):e356. doi: 10.1002/ctm2.356. Clin Transl Med. 2021. PMID: 33783998 Free PMC article. Review.
-
Unbiased Single-Cell Sequencing of Hematopoietic and Immune Cells from Aplastic Anemia Reveals the Contributors of Hematopoiesis Failure and Dysfunctional Immune Regulation.Adv Sci (Weinh). 2024 Mar;11(10):e2304539. doi: 10.1002/advs.202304539. Epub 2023 Dec 25. Adv Sci (Weinh). 2024. PMID: 38145351 Free PMC article.
-
15-Keto prostaglandin E2 suppresses STAT3 signaling and inhibits breast cancer cell growth and progression.Redox Biol. 2019 May;23:101175. doi: 10.1016/j.redox.2019.101175. Epub 2019 Mar 28. Redox Biol. 2019. PMID: 31129031 Free PMC article.
References
-
- Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA, et al. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65(3):982–90. - PubMed
-
- Yamaue H, Satoi S, Kanbe T, Miyazawa M, Tani M, Kawai M, et al. Phase II clinical study of alternate-day oral therapy with S-1 as first-line chemotherapy for locally advanced and metastatic pancreatic cancer. Cancer Chemother Pharmacol. 2013. - PubMed
-
- Chu J, Lloyd FL, Trifan OC, Knapp B, Rizzo MT. Potential involvement of the cyclooxygenase-2 pathway in the regulation of tumor-associated angiogenesis and growth in pancreatic cancer. Mol Cancer Ther. 2003;2(1):1–7. - PubMed
-
- Chou WL, Chuang LM, Chou CC, Wang AH, Lawson JA, FitzGerald GA, et al. Identification of a novel prostaglandin reductase reveals the involvement of prostaglandin E2 catabolism in regulation of peroxisome proliferator-activated receptor gamma activation. J Biol Chem. 2007;282(25):18162–72. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
