

Reduction of aberrant NF-κB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice
- PMID: 26821816
- PMCID: PMC4740176
- DOI: 10.1038/ncomms10520
Reduction of aberrant NF-κB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice
Abstract
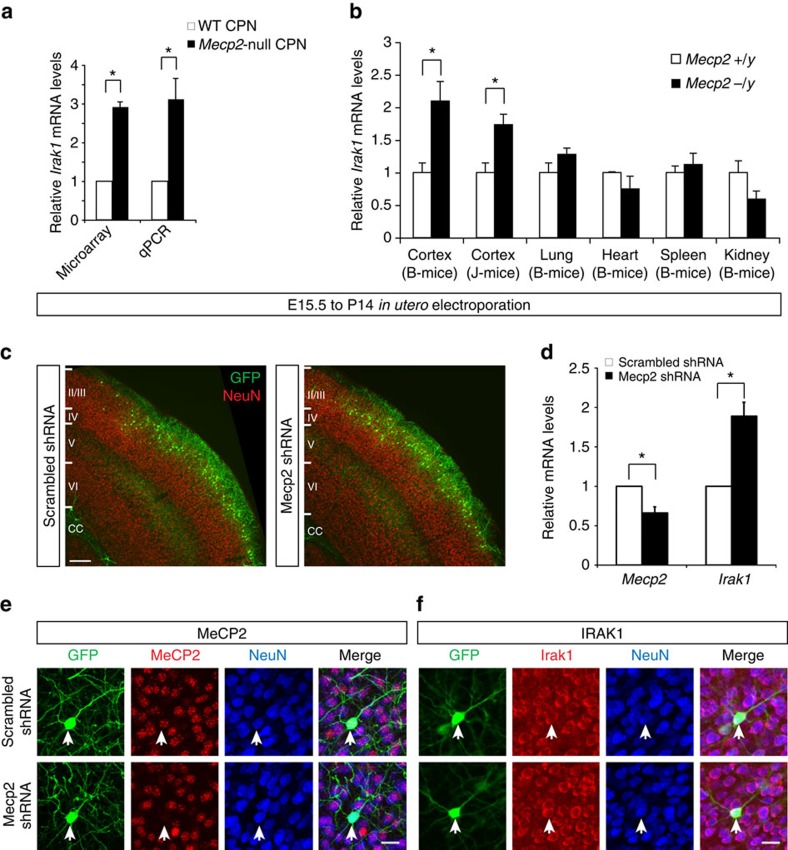
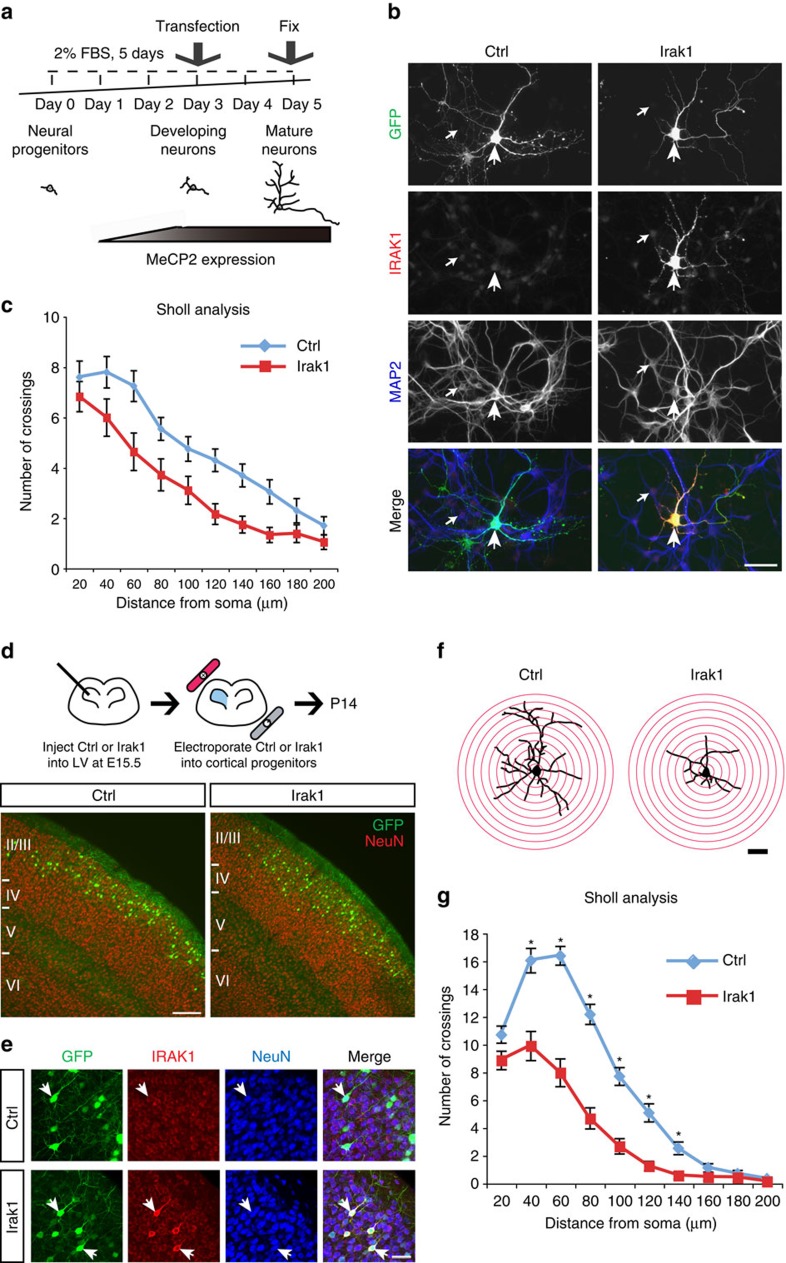
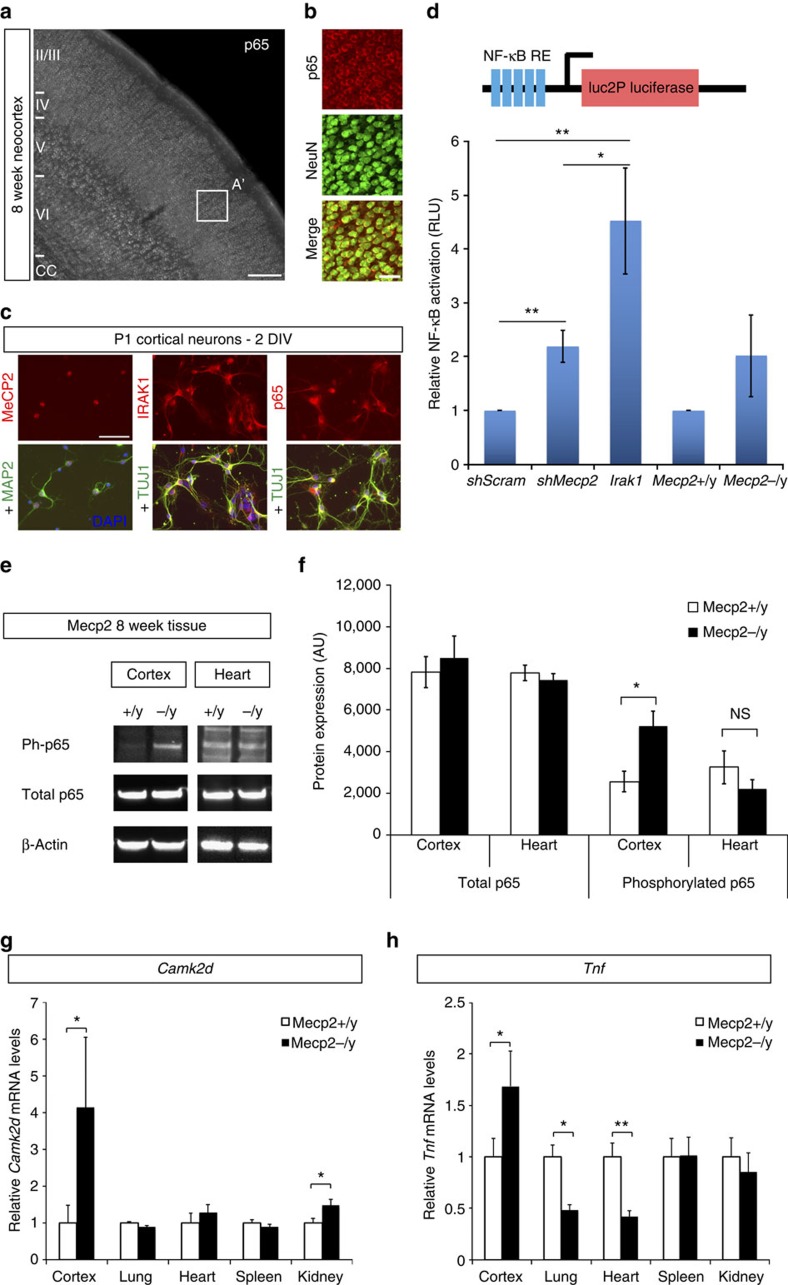
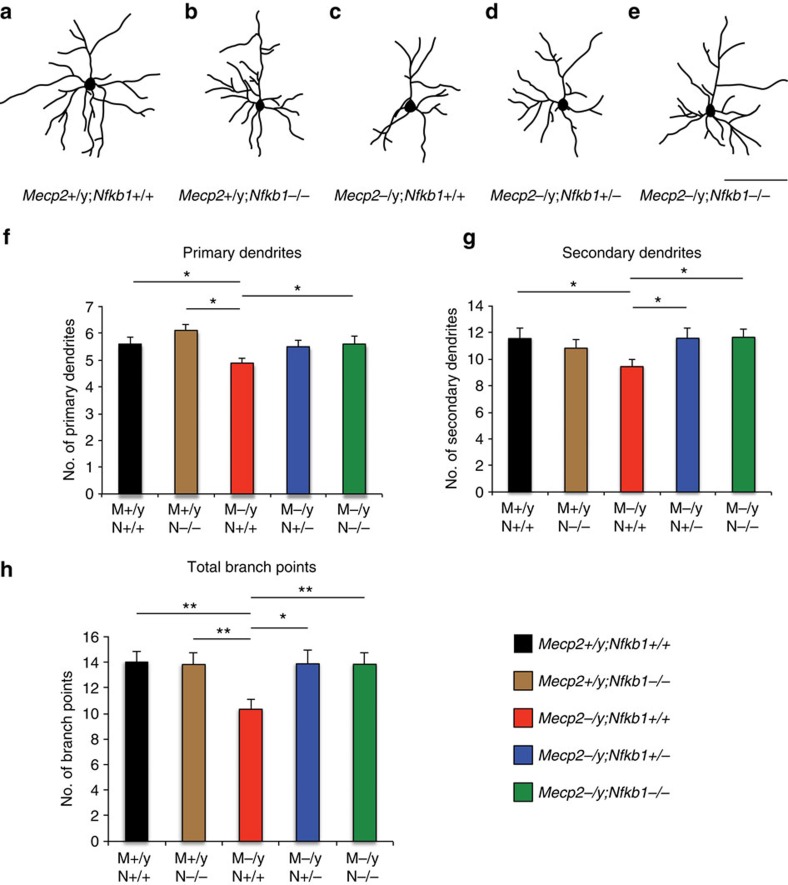
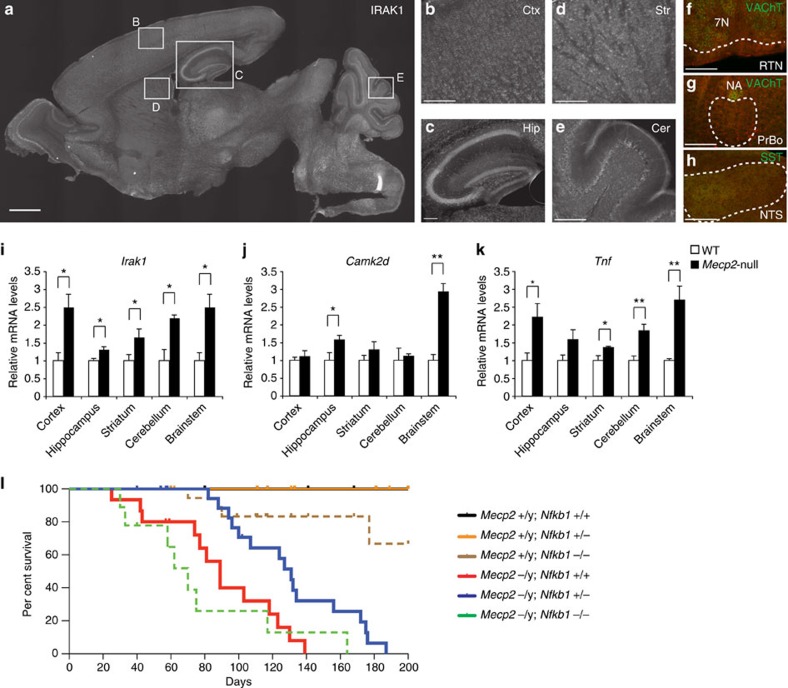
Mutations in the transcriptional regulator Mecp2 cause the severe X-linked neurodevelopmental disorder Rett syndrome (RTT). In this study, we investigate genes that function downstream of MeCP2 in cerebral cortex circuitry, and identify upregulation of Irak1, a central component of the NF-κB pathway. We show that overexpression of Irak1 mimics the reduced dendritic complexity of Mecp2-null cortical callosal projection neurons (CPN), and that NF-κB signalling is upregulated in the cortex with Mecp2 loss-of-function. Strikingly, we find that genetically reducing NF-κB signalling in Mecp2-null mice not only ameliorates CPN dendritic complexity but also substantially extends their normally shortened lifespan, indicating broader roles for NF-κB signalling in RTT pathogenesis. These results provide new insight into both the fundamental neurobiology of RTT, and potential therapeutic strategies via NF-κB pathway modulation.
Figures
References
-
- Chahrour M. & Zoghbi H. Y. The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437 (2007) . - PubMed
-
- Amir R. E. et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188 (1999) . - PubMed
-
- Chen R. Z., Akbarian S., Tudor M. & Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331 (2001) . - PubMed
-
- Guy J., Hendrich B., Holmes M., Martin J. E. & Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326 (2001) . - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
