

Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca(2+)-dependent inactivation
- PMID: 26822303
- PMCID: PMC4740114
- DOI: 10.1038/ncomms10370
Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca(2+)-dependent inactivation
Abstract
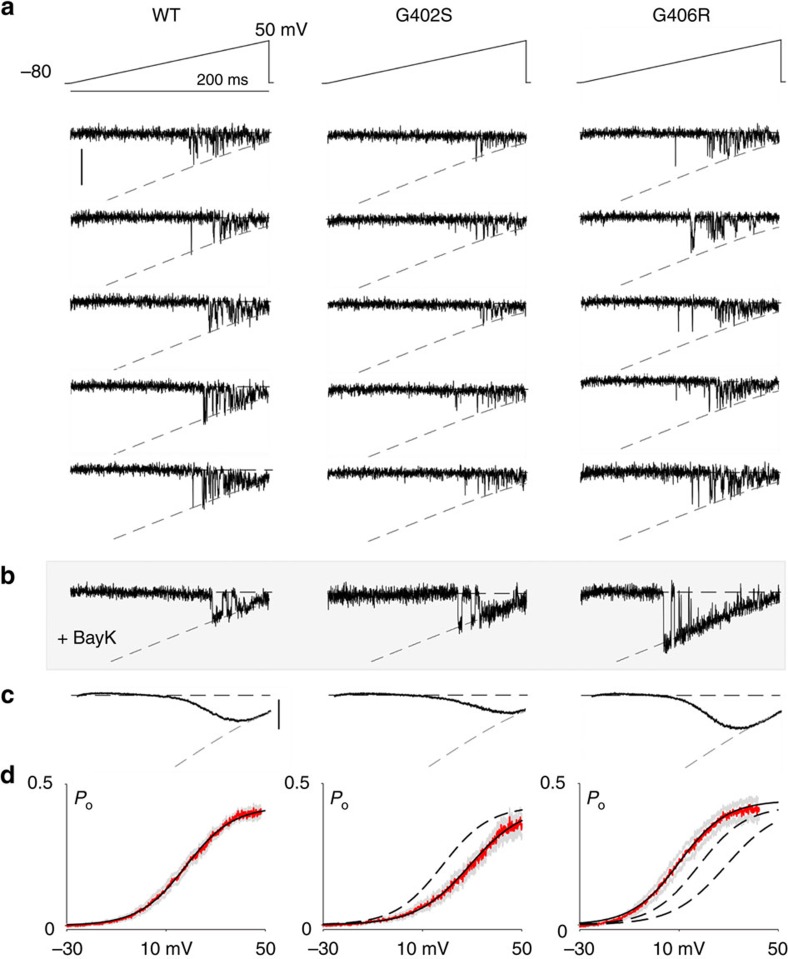
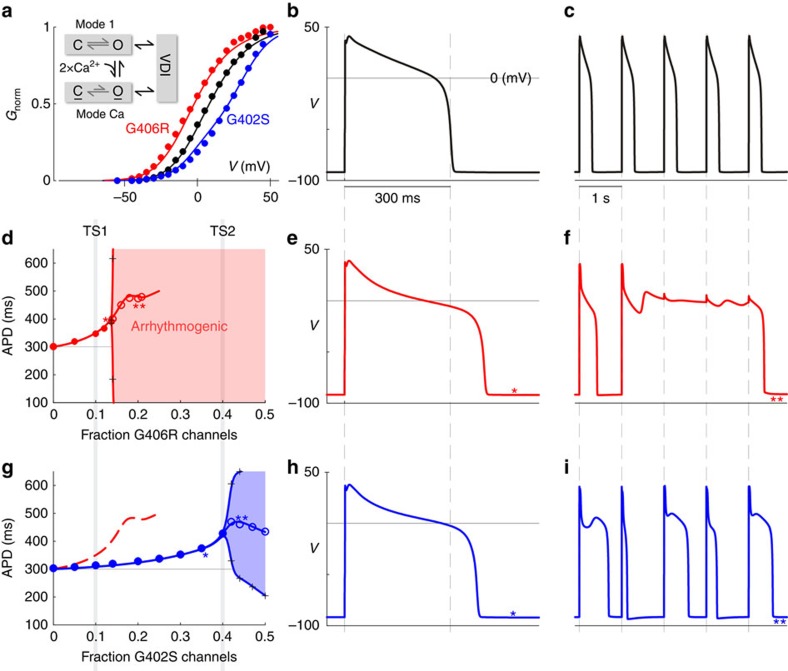
Timothy Syndrome (TS) is a multisystem disorder, prominently featuring cardiac action potential prolongation with paroxysms of life-threatening arrhythmias. The underlying defect is a single de novo missense mutation in CaV1.2 channels, either G406R or G402S. Notably, these mutations are often viewed as equivalent, as they produce comparable defects in voltage-dependent inactivation and cause similar manifestations in patients. Yet, their effects on calcium-dependent inactivation (CDI) have remained uncertain. Here, we find a significant defect in CDI in TS channels, and uncover a remarkable divergence in the underlying mechanism for G406R versus G402S variants. Moreover, expression of these TS channels in cultured adult guinea pig myocytes, combined with a quantitative ventricular myocyte model, reveals a threshold behaviour in the induction of arrhythmias due to TS channel expression, suggesting an important therapeutic principle: a small shift in the complement of mutant versus wild-type channels may confer significant clinical improvement.
Figures





References
-
- Splawski I. et al.. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004) . - PubMed
-
- Liao P., Yong T. F., Liang M. C., Yue D. T. & Soong T. W. Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc. Res. 68, 197–203 (2005) . - PubMed
-
- Liao P. & Soong T. W. CaV1.2 channelopathies: from arrhythmias to autism, bipolar disorder, and immunodeficiency. Pflugers Arch. 460, 353–359 (2010) . - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
