

2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition
- PMID: 26822609
- PMCID: PMC6946184
- DOI: 10.1126/science.aad7974
2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition
Abstract
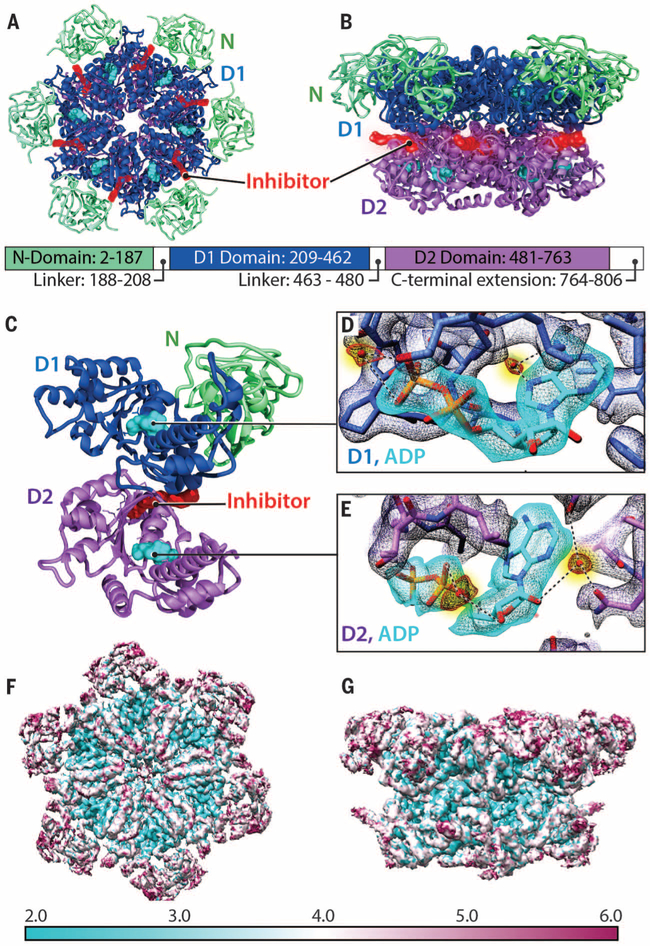
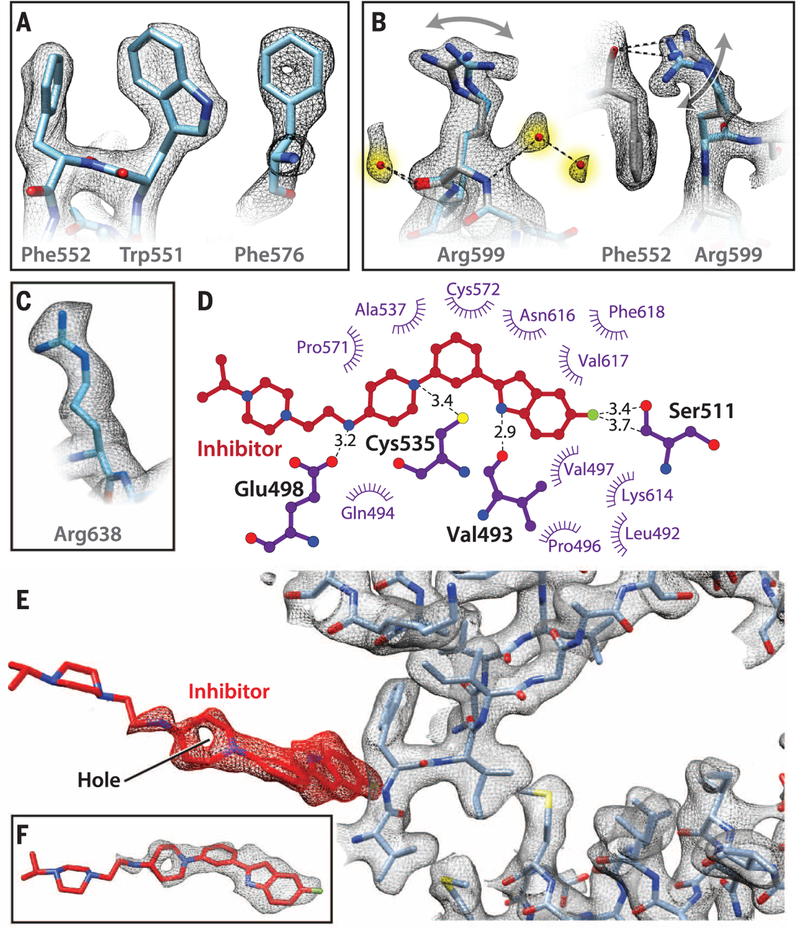
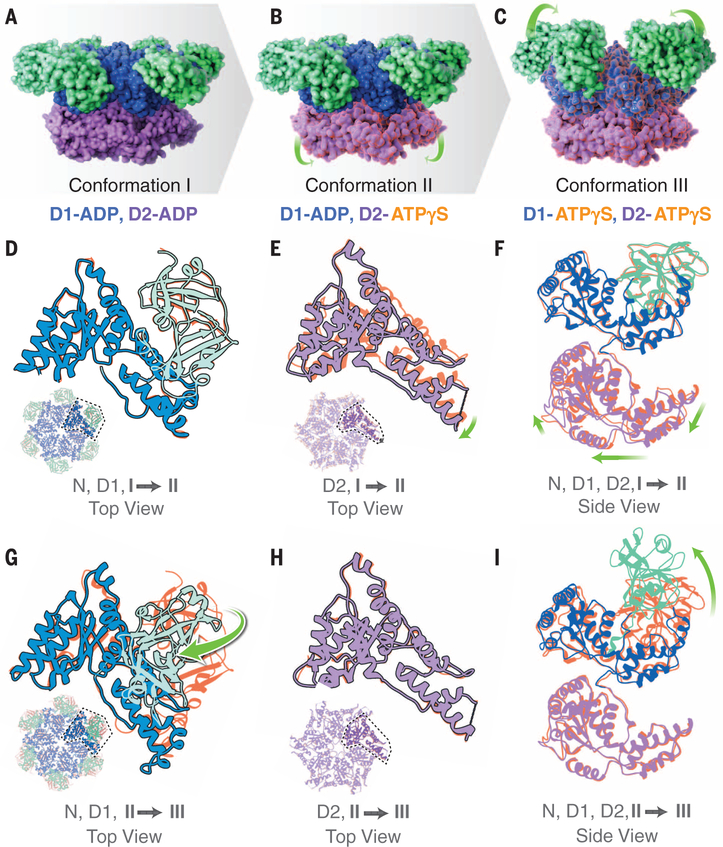
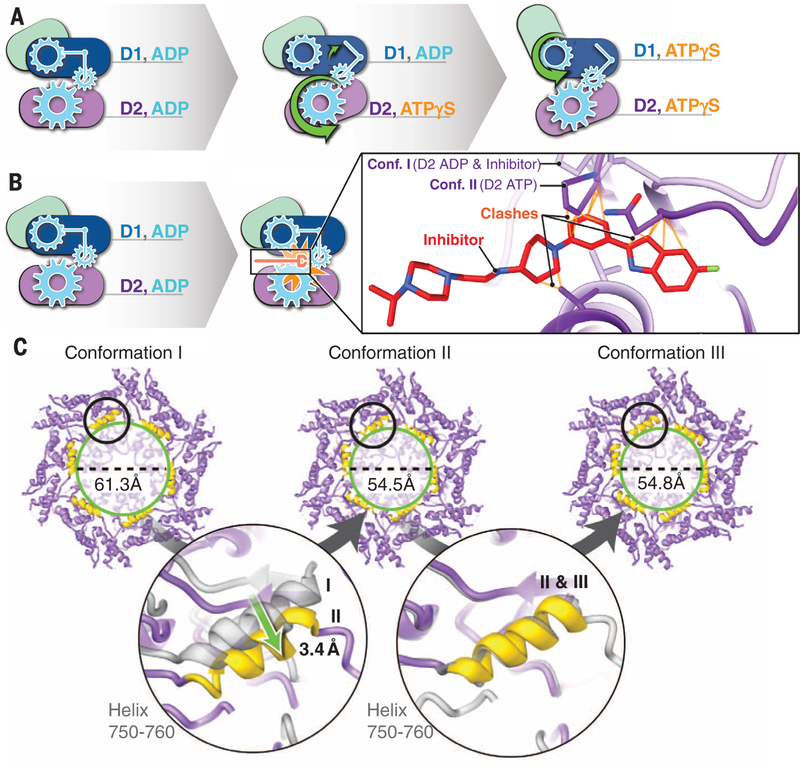
p97 is a hexameric AAA+ adenosine triphosphatase (ATPase) that is an attractive target for cancer drug development. We report cryo-electron microscopy (cryo-EM) structures for adenosine diphosphate (ADP)-bound, full-length, hexameric wild-type p97 in the presence and absence of an allosteric inhibitor at resolutions of 2.3 and 2.4 angstroms, respectively. We also report cryo-EM structures (at resolutions of ~3.3, 3.2, and 3.3 angstroms, respectively) for three distinct, coexisting functional states of p97 with occupancies of zero, one, or two molecules of adenosine 5'-O-(3-thiotriphosphate) (ATPγS) per protomer. A large corkscrew-like change in molecular architecture, coupled with upward displacement of the N-terminal domain, is observed only when ATPγS is bound to both the D1 and D2 domains of the protomer. These cryo-EM structures establish the sequence of nucleotide-driven structural changes in p97 at atomic resolution. They also enable elucidation of the binding mode of an allosteric small-molecule inhibitor to p97 and illustrate how inhibitor binding at the interface between the D1 and D2 domains prevents propagation of the conformational changes necessary for p97 function.
Copyright © 2016, American Association for the Advancement of Science.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
