

Pluralistic and stochastic gene regulation: examples, models and consistent theory
- PMID: 26823500
- PMCID: PMC4889914
- DOI: 10.1093/nar/gkw042
Pluralistic and stochastic gene regulation: examples, models and consistent theory
Abstract
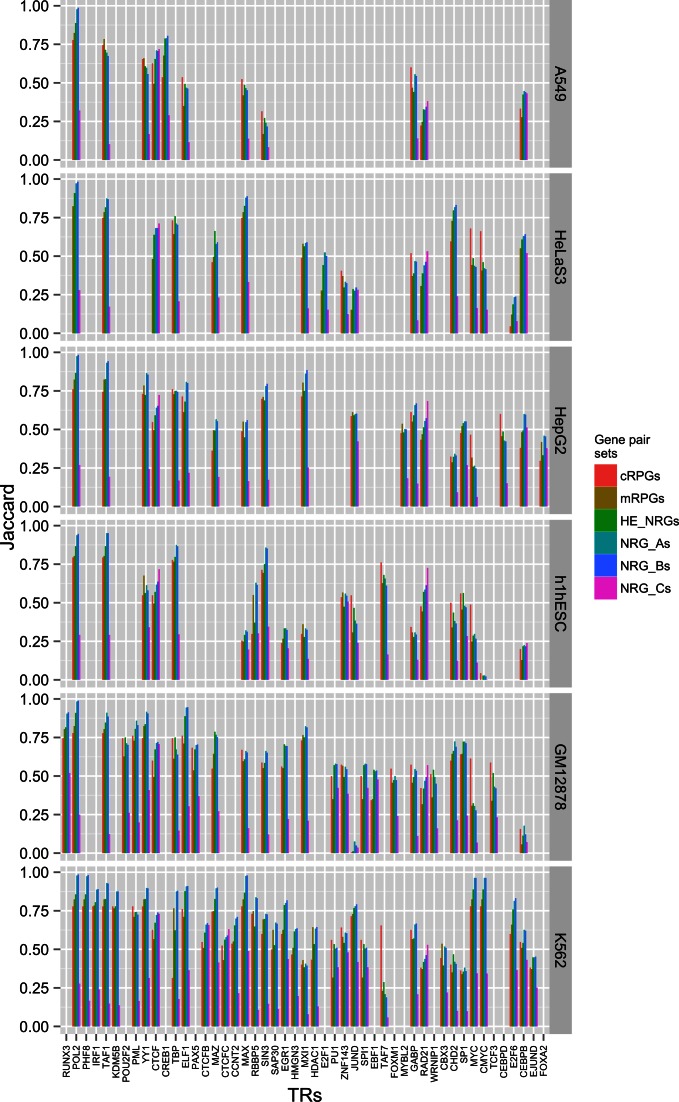
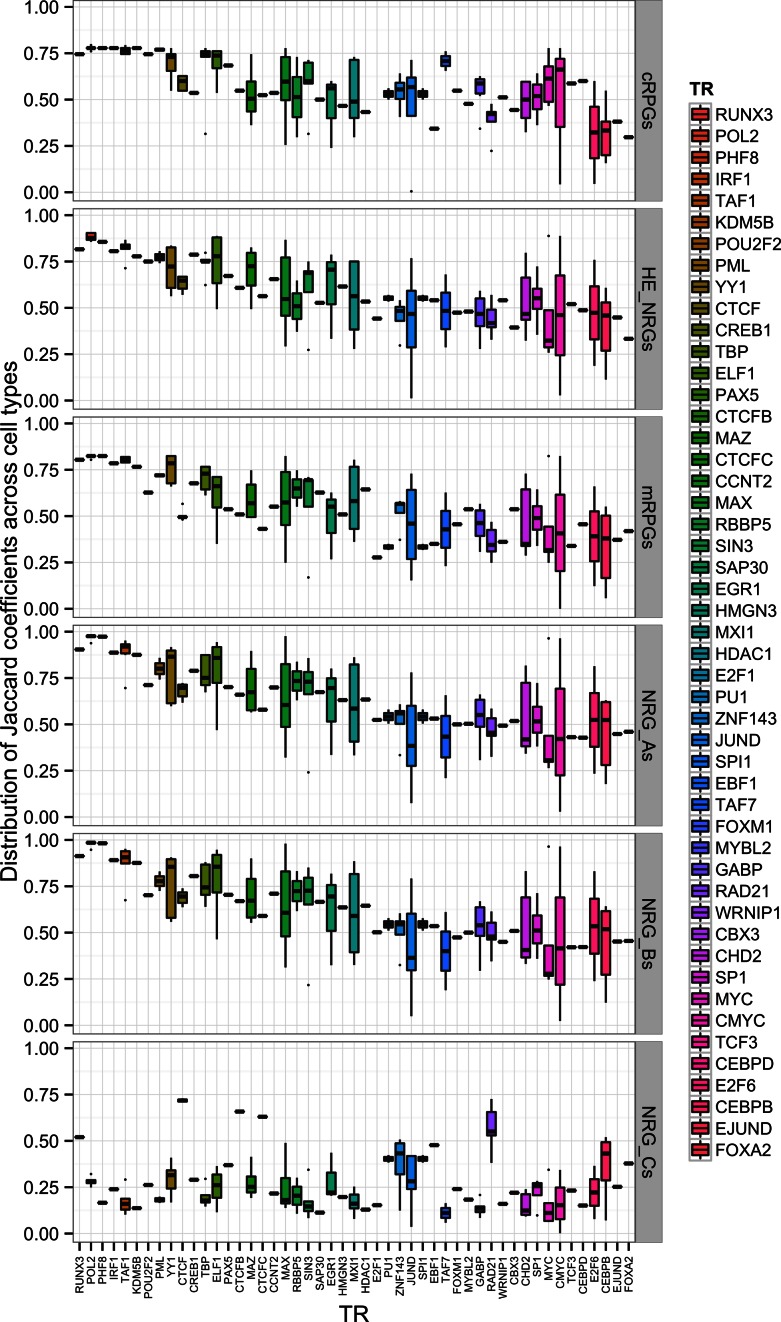
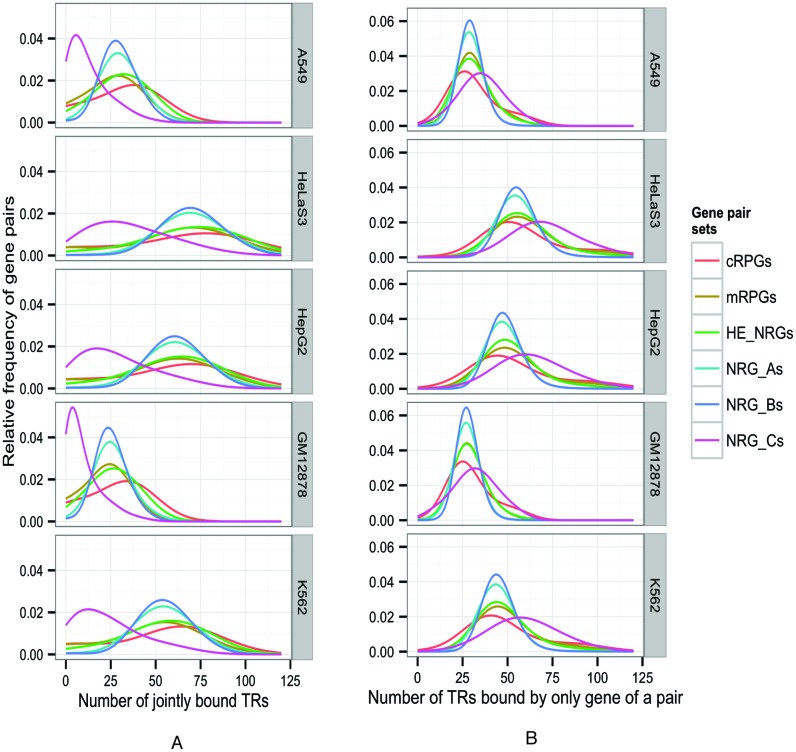
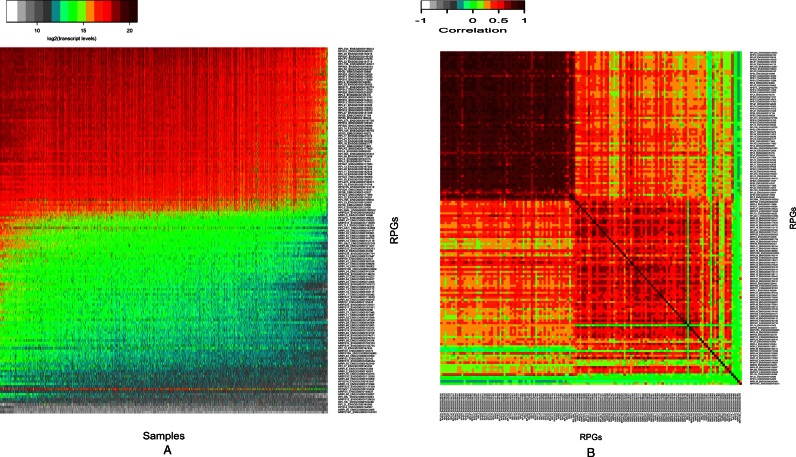
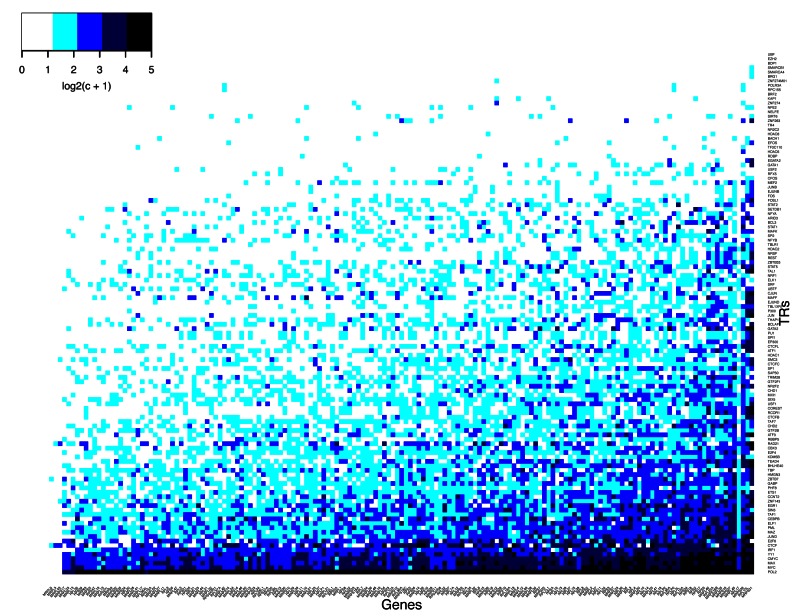
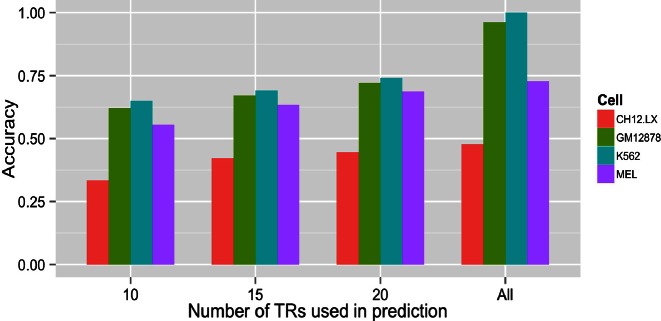
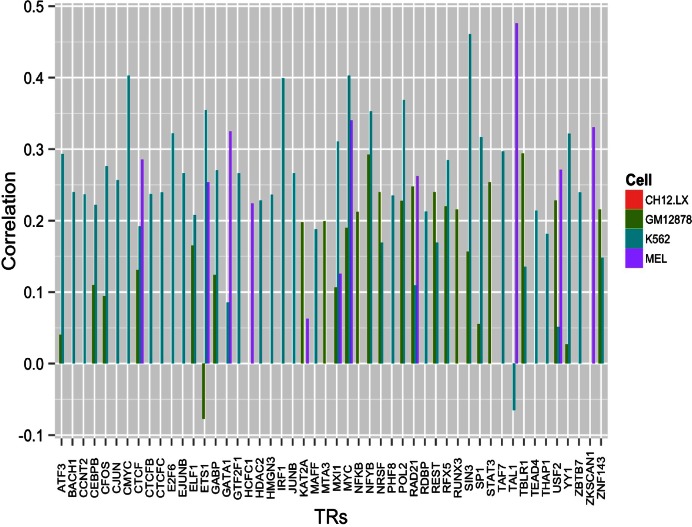
We present a theory of pluralistic and stochastic gene regulation. To bridge the gap between empirical studies and mathematical models, we integrate pre-existing observations with our meta-analyses of the ENCODE ChIP-Seq experiments. Earlier evidence includes fluctuations in levels, location, activity, and binding of transcription factors, variable DNA motifs, and bursts in gene expression. Stochastic regulation is also indicated by frequently subdued effects of knockout mutants of regulators, their evolutionary losses/gains and massive rewiring of regulatory sites. We report wide-spread pluralistic regulation in ≈800 000 tightly co-expressed pairs of diverse human genes. Typically, half of ≈50 observed regulators bind to both genes reproducibly, twice more than in independently expressed gene pairs. We also examine the largest set of co-expressed genes, which code for cytoplasmic ribosomal proteins. Numerous regulatory complexes are highly significant enriched in ribosomal genes compared to highly expressed non-ribosomal genes. We could not find any DNA-associated, strict sense master regulator. Despite major fluctuations in transcription factor binding, our machine learning model accurately predicted transcript levels using binding sites of 20+ regulators. Our pluralistic and stochastic theory is consistent with partially random binding patterns, redundancy, stochastic regulator binding, burst-like expression, degeneracy of binding motifs and massive regulatory rewiring during evolution.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures


Similar articles
-
A resource for characterizing genome-wide binding and putative target genes of transcription factors expressed during secondary growth and wood formation in Populus.Plant J. 2015 Jun;82(5):887-98. doi: 10.1111/tpj.12850. Plant J. 2015. PMID: 25903933
-
Using Weeder, Pscan, and PscanChIP for the Discovery of Enriched Transcription Factor Binding Site Motifs in Nucleotide Sequences.Curr Protoc Bioinformatics. 2014 Sep 8;47:2.11.1-31. doi: 10.1002/0471250953.bi0211s47. Curr Protoc Bioinformatics. 2014. PMID: 25199791
-
MOCCS: Clarifying DNA-binding motif ambiguity using ChIP-Seq data.Comput Biol Chem. 2016 Aug;63:62-72. doi: 10.1016/j.compbiolchem.2016.01.014. Epub 2016 Feb 13. Comput Biol Chem. 2016. PMID: 26971251
-
Utility of next-generation RNA-sequencing in identifying chimeric transcription involving human endogenous retroviruses.APMIS. 2016 Jan-Feb;124(1-2):127-39. doi: 10.1111/apm.12477. APMIS. 2016. PMID: 26818267 Review.
-
The rewiring of transcription circuits in evolution.Curr Opin Genet Dev. 2017 Dec;47:121-127. doi: 10.1016/j.gde.2017.09.004. Epub 2017 Nov 8. Curr Opin Genet Dev. 2017. PMID: 29120735 Free PMC article. Review.
Cited by
-
Neurog3-Independent Methylation Is the Earliest Detectable Mark Distinguishing Pancreatic Progenitor Identity.Dev Cell. 2019 Jan 7;48(1):49-63.e7. doi: 10.1016/j.devcel.2018.11.048. Dev Cell. 2019. PMID: 30620902 Free PMC article.
-
Dynamic and Modularized MicroRNA Regulation and Its Implication in Human Cancers.Sci Rep. 2017 Oct 17;7(1):13356. doi: 10.1038/s41598-017-13470-5. Sci Rep. 2017. PMID: 29042600 Free PMC article.
References
-
- Montgomery S.B., Dermitzakis E.T. From expression QTLs to personalized transcriptomics. Nat. Rev. Genet. 2011;12:277–282. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
