

Predicted Mutation Strength of Nontruncating PKD1 Mutations Aids Genotype-Phenotype Correlations in Autosomal Dominant Polycystic Kidney Disease
- PMID: 26823553
- PMCID: PMC5004648
- DOI: 10.1681/ASN.2015050583
Predicted Mutation Strength of Nontruncating PKD1 Mutations Aids Genotype-Phenotype Correlations in Autosomal Dominant Polycystic Kidney Disease
Abstract
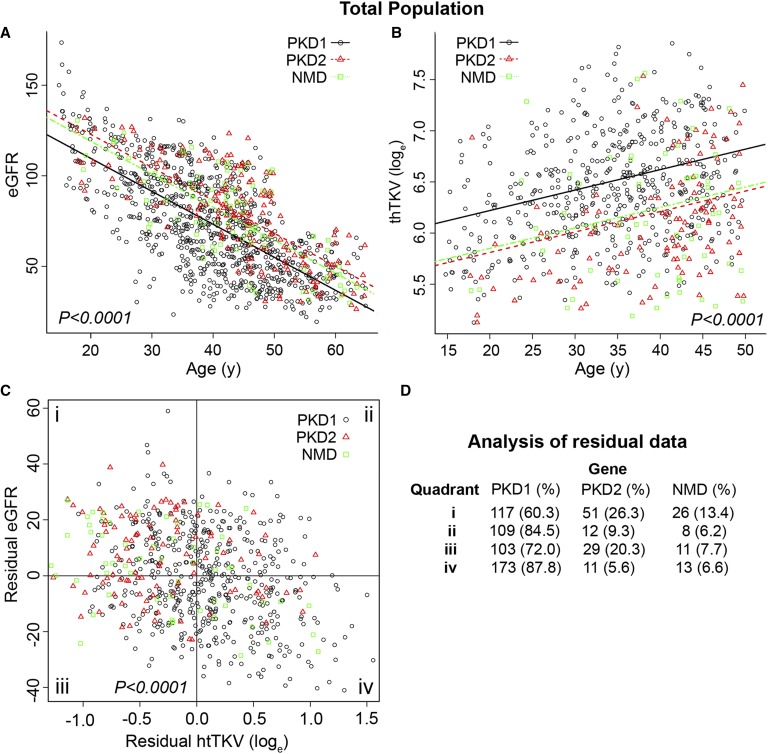
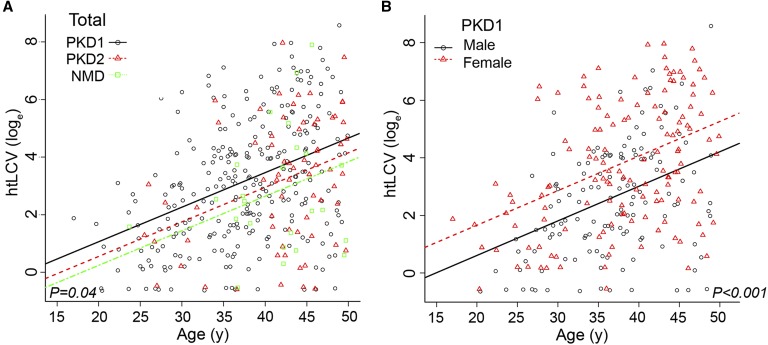
Autosomal dominant polycystic kidney disease (ADPKD) often results in ESRD but with a highly variable course. Mutations to PKD1 or PKD2 cause ADPKD; both loci have high levels of allelic heterogeneity. We evaluated genotype-phenotype correlations in 1119 patients (945 families) from the HALT Progression of PKD Study and the Consortium of Radiologic Imaging Study of PKD Study. The population was defined as: 77.7% PKD1, 14.7% PKD2, and 7.6% with no mutation detected (NMD). Phenotypic end points were sex, eGFR, height-adjusted total kidney volume (htTKV), and liver cyst volume. Analysis of the eGFR and htTKV measures showed that the PKD1 group had more severe disease than the PKD2 group, whereas the NMD group had a PKD2-like phenotype. In both the PKD1 and PKD2 populations, men had more severe renal disease, but women had larger liver cyst volumes. Compared with nontruncating PKD1 mutations, truncating PKD1 mutations associated with lower eGFR, but the mutation groups were not differentiated by htTKV. PKD1 nontruncating mutations were evaluated for conservation and chemical change and subdivided into strong (mutation strength group 2 [MSG2]) and weak (MSG3) mutation groups. Analysis of eGFR and htTKV measures showed that patients with MSG3 but not MSG2 mutations had significantly milder disease than patients with truncating cases (MSG1), an association especially evident in extreme decile populations. Overall, we have quantified the contribution of genic and PKD1 allelic effects and sex to the ADPKD phenotype. Intrafamilial correlation analysis showed that other factors shared by families influence htTKV, with these additional genetic/environmental factors significantly affecting the ADPKD phenotype.
Keywords: ADPKD; Genotype/phenotype; Prognostic studies.
Copyright © 2016 by the American Society of Nephrology.
Figures



References
-
- Torres VE, Harris PC, Pirson Y: Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007 - PubMed
-
- Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM, Walker DL, Bae K, Zhang QJ, Thompson PA, Miller JP, Harris PC CRISP Consortium : Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 18: 2143–2160, 2007 - PubMed
-
- Audrézet MP, Cornec-Le Gall E, Chen JM, Redon S, Quéré I, Creff J, Bénech C, Maestri S, Le Meur Y, Férec C: Autosomal dominant polycystic kidney disease: Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat 33: 1239–1250, 2012 - PubMed
-
- Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D: Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 353: 103–107, 1999 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK062401/DK/NIDDK NIH HHS/United States
- M01 RR000585/RR/NCRR NIH HHS/United States
- U01 DK056956/DK/NIDDK NIH HHS/United States
- P30 DK090728/DK/NIDDK NIH HHS/United States
- U01 DK062402/DK/NIDDK NIH HHS/United States
- UL1 RR025780/RR/NCRR NIH HHS/United States
- UL1 TR001064/TR/NCATS NIH HHS/United States
- UL1 RR025752/RR/NCRR NIH HHS/United States
- U01 DK056957/DK/NIDDK NIH HHS/United States
- M01 RR000039/RR/NCRR NIH HHS/United States
- UL1 RR033179/RR/NCRR NIH HHS/United States
- P30 DK106912/DK/NIDDK NIH HHS/United States
- UL1 TR000001/TR/NCATS NIH HHS/United States
- M01 RR001032/RR/NCRR NIH HHS/United States
- M01 RR000054/RR/NCRR NIH HHS/United States
- M01 RR023940/RR/NCRR NIH HHS/United States
- UL1 TR000135/TR/NCATS NIH HHS/United States
- U01 DK056943/DK/NIDDK NIH HHS/United States
- U01 DK062408/DK/NIDDK NIH HHS/United States
- UL1 TR000454/TR/NCATS NIH HHS/United States
- U01 DK056961/DK/NIDDK NIH HHS/United States
- M01 RR000051/RR/NCRR NIH HHS/United States
- U01 DK062411/DK/NIDDK NIH HHS/United States
- R01 DK044863/DK/NIDDK NIH HHS/United States
- UL1 TR001082/TR/NCATS NIH HHS/United States
- UL1 TR001102/TR/NCATS NIH HHS/United States
- U01 DK082230/DK/NIDDK NIH HHS/United States
- UL1 RR024150/RR/NCRR NIH HHS/United States
- UL1 RR025758/RR/NCRR NIH HHS/United States
- UL1 RR025008/RR/NCRR NIH HHS/United States
- U01 DK062410/DK/NIDDK NIH HHS/United States
- R01 DK058816/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
