

Targeting NF-κB in glioblastoma: A therapeutic approach
- PMID: 26824076
- PMCID: PMC4730860
- DOI: 10.1126/sciadv.1501292
Targeting NF-κB in glioblastoma: A therapeutic approach
Abstract
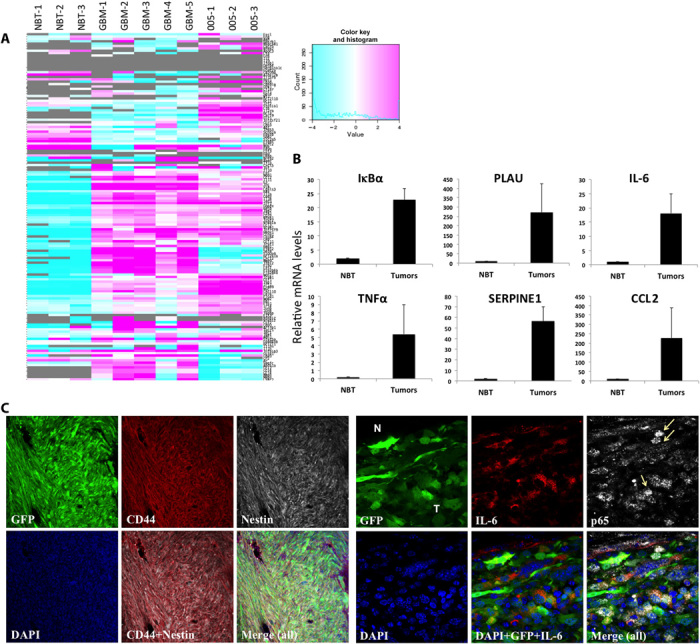
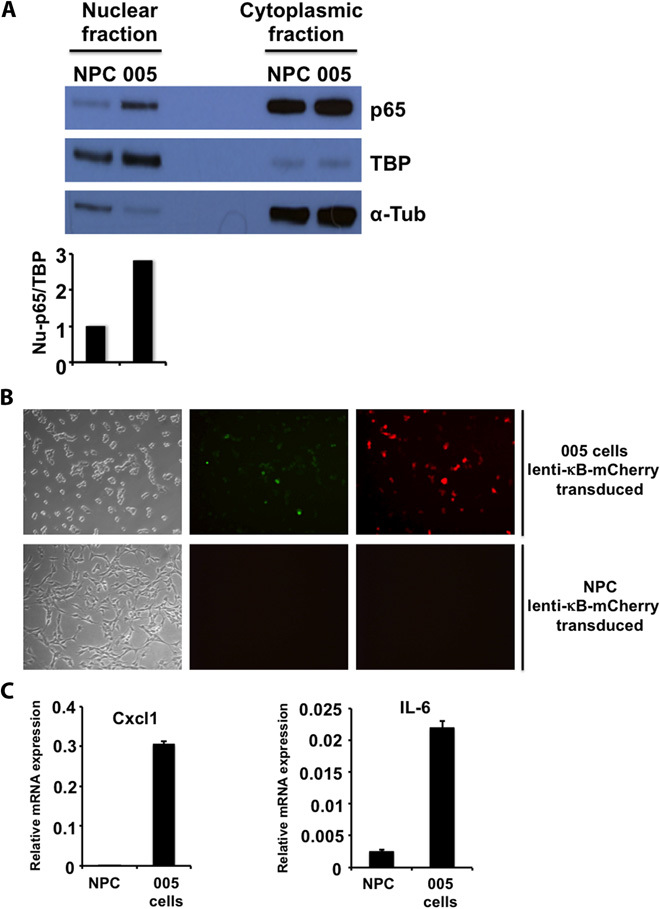
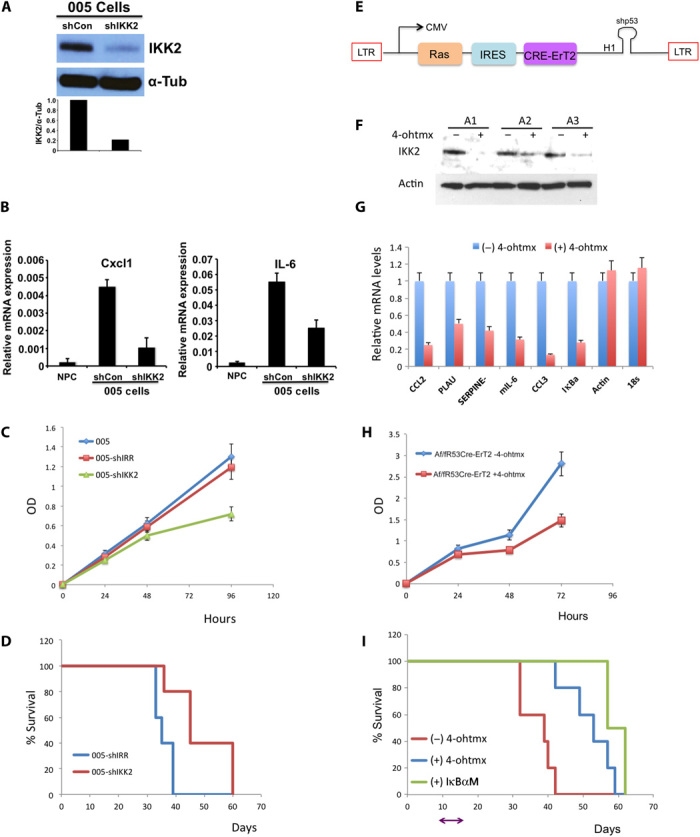
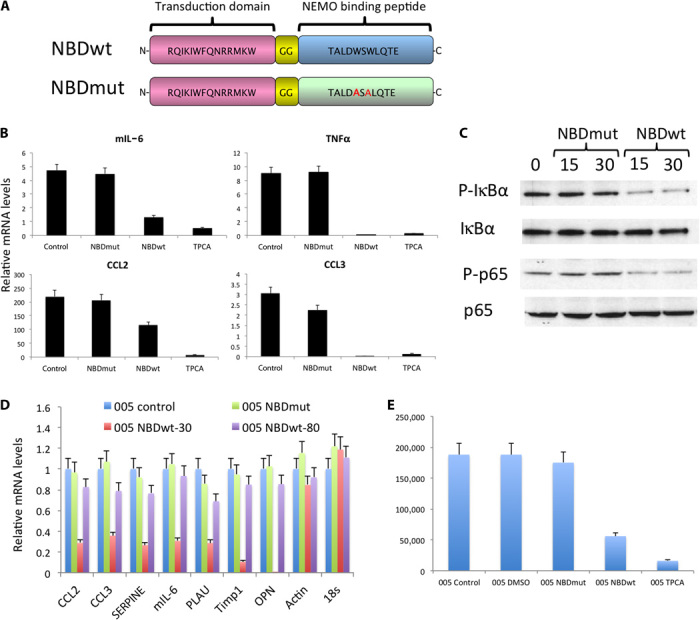
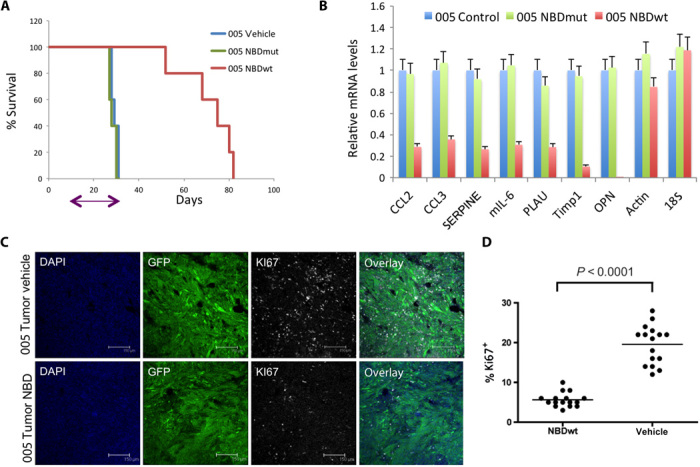
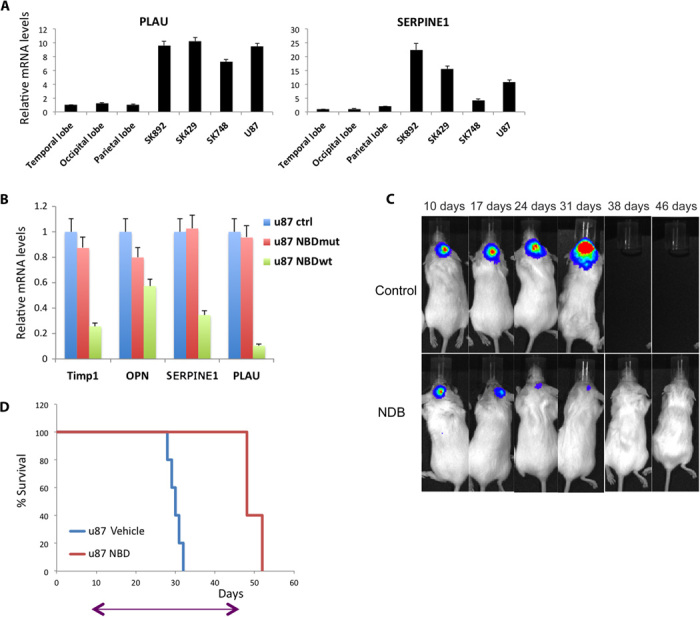
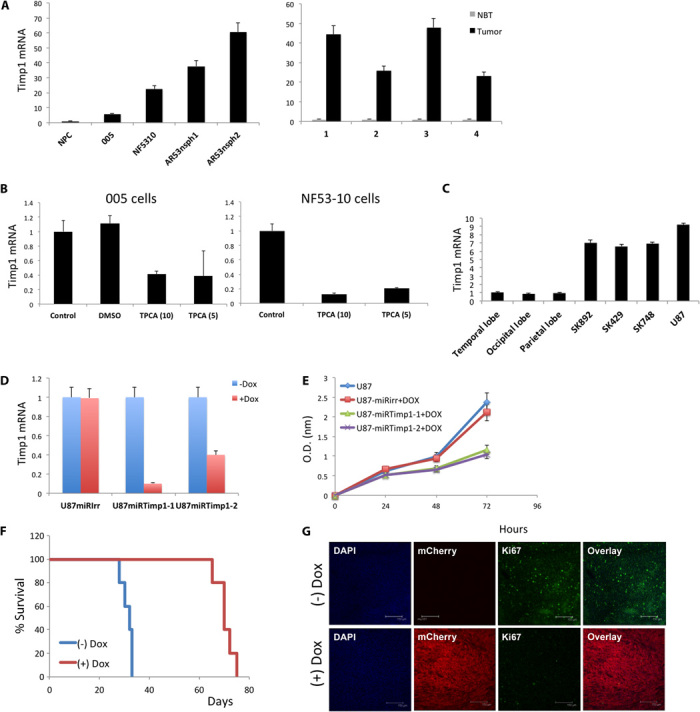
Glioblastoma multiforme (GBM) is the most common and lethal form of intracranial tumor. We have established a lentivirus-induced mouse model of malignant gliomas, which faithfully captures the pathophysiology and molecular signature of mesenchymal human GBM. RNA-Seq analysis of these tumors revealed high nuclear factor κB (NF-κB) activation showing enrichment of known NF-κB target genes. Inhibition of NF-κB by either depletion of IκB kinase 2 (IKK2), expression of a IκBαM super repressor, or using a NEMO (NF-κB essential modifier)-binding domain (NBD) peptide in tumor-derived cell lines attenuated tumor proliferation and prolonged mouse survival. Timp1, one of the NF-κB target genes significantly up-regulated in GBM, was identified to play a role in tumor proliferation and growth. Inhibition of NF-κB activity or silencing of Timp1 resulted in slower tumor growth in both mouse and human GBM models. Our results suggest that inhibition of NF-κB activity or targeting of inducible NF-κB genes is an attractive therapeutic approach for GBM.
Keywords: Gliomas; NF-κB inhibitors; TIMP1; Xenografts.
Figures
References
-
- Furnari F. B., Fenton T., Bachoo R. M., Mukasa A., Stommel J. M., Stegh A., Hahn W. C., Ligon K. L., Louis D. N., Brennan C., Chin L., DePinho R. A., Cavenee W. K., Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 21, 2683–2710 (2007). - PubMed
-
- Huse J. T., Holland E. C., Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 10, 319–331 (2010). - PubMed
-
- Stiles C. D., Rowitch D. H., Glioma stem cells: A midterm exam. Neuron 58, 832–846 (2008). - PubMed
-
- Cooper L. A. D., Kong J., Gutman D. A., Wang F., Cholleti S. R., Pan T. C., Widener P. M., Sharma A., Mikkelsen T., Flanders A. E., Rubin D. L., Van Meir E. G., Kurc T. M., Moreno C. S., Brat D. J., Saltz J. H., An integrative approach for in silico glioma research. IEEE Trans. Biomed. Eng. 57, 2617–2621 (2010). - PMC - PubMed
-
- Huse J. T., Phillips H. S., Brennan C. W., Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 59, 1190–1199 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
