

Overcoming potential energy distortions in constrained internal coordinate molecular dynamics simulations
- PMID: 26827207
- PMCID: PMC4733083
- DOI: 10.1063/1.4939532
Overcoming potential energy distortions in constrained internal coordinate molecular dynamics simulations
Abstract
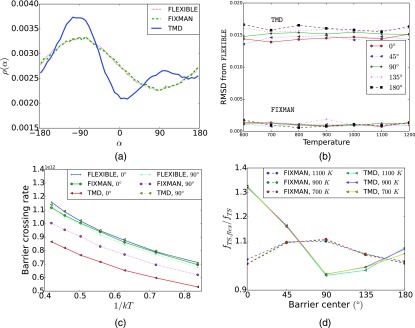
The Internal Coordinate Molecular Dynamics (ICMD) method is an attractive molecular dynamics (MD) method for studying the dynamics of bonded systems such as proteins and polymers. It offers a simple venue for coarsening the dynamics model of a system at multiple hierarchical levels. For example, large scale protein dynamics can be studied using torsional dynamics, where large domains or helical structures can be treated as rigid bodies and the loops connecting them as flexible torsions. ICMD with such a dynamic model of the protein, combined with enhanced conformational sampling method such as temperature replica exchange, allows the sampling of large scale domain motion involving high energy barrier transitions. Once these large scale conformational transitions are sampled, all-torsion, or even all-atom, MD simulations can be carried out for the low energy conformations sampled via coarse grained ICMD to calculate the energetics of distinct conformations. Such hierarchical MD simulations can be carried out with standard all-atom forcefields without the need for compromising on the accuracy of the forces. Using constraints to treat bond lengths and bond angles as rigid can, however, distort the potential energy landscape of the system and reduce the number of dihedral transitions as well as conformational sampling. We present here a two-part solution to overcome such distortions of the potential energy landscape with ICMD models. To alleviate the intrinsic distortion that stems from the reduced phase space in torsional MD, we use the Fixman compensating potential. To additionally alleviate the extrinsic distortion that arises from the coupling between the dihedral angles and bond angles within a force field, we propose a hybrid ICMD method that allows the selective relaxing of bond angles. This hybrid ICMD method bridges the gap between all-atom MD and torsional MD. We demonstrate with examples that these methods together offer a solution to eliminate the potential energy distortions encountered in constrained ICMD simulations of peptide molecules.
Figures












Similar articles
-
Internal coordinate molecular dynamics: a foundation for multiscale dynamics.J Phys Chem B. 2015 Jan 29;119(4):1233-42. doi: 10.1021/jp509136y. Epub 2015 Jan 6. J Phys Chem B. 2015. PMID: 25517406 Free PMC article.
-
GneimoSim: a modular internal coordinates molecular dynamics simulation package.J Comput Chem. 2014 Dec 5;35(31):2245-55. doi: 10.1002/jcc.23743. Epub 2014 Sep 27. J Comput Chem. 2014. PMID: 25263538 Free PMC article.
-
Advanced techniques for constrained internal coordinate molecular dynamics.J Comput Chem. 2013 Apr 30;34(11):904-14. doi: 10.1002/jcc.23200. Epub 2013 Jan 23. J Comput Chem. 2013. PMID: 23345138 Free PMC article.
-
Multiscale molecular dynamics simulations of rotary motor proteins.Biophys Rev. 2018 Apr;10(2):605-615. doi: 10.1007/s12551-017-0373-4. Epub 2017 Dec 4. Biophys Rev. 2018. PMID: 29204882 Free PMC article. Review.
-
Detecting Functional Dynamics in Proteins with Comparative Perturbed-Ensembles Analysis.Acc Chem Res. 2019 Dec 17;52(12):3455-3464. doi: 10.1021/acs.accounts.9b00485. Epub 2019 Dec 3. Acc Chem Res. 2019. PMID: 31793290 Review.
Cited by
-
Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins.Biomolecules. 2019 Apr 11;9(4):146. doi: 10.3390/biom9040146. Biomolecules. 2019. PMID: 30979035 Free PMC article. Review.
-
Hamiltonian Monte Carlo with Constrained Molecular Dynamics as Gibbs Sampling.J Chem Theory Comput. 2017 Oct 10;13(10):4649-4659. doi: 10.1021/acs.jctc.7b00570. Epub 2017 Sep 27. J Chem Theory Comput. 2017. PMID: 28892630 Free PMC article.
-
Comparison of the United- and All-Atom Representations of (Halo)alkanes Based on Two Condensed-Phase Force Fields Optimized against the Same Experimental Data Set.J Chem Theory Comput. 2022 Nov 8;18(11):6757-6778. doi: 10.1021/acs.jctc.2c00524. Epub 2022 Oct 3. J Chem Theory Comput. 2022. PMID: 36190354 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous