

Maximally asymmetric transbilayer distribution of anionic lipids alters the structure and interaction with lipids of an amyloidogenic protein dimer bound to the membrane surface
- PMID: 26827904
- PMCID: PMC4822834
- DOI: 10.1016/j.chemphyslip.2016.01.002
Maximally asymmetric transbilayer distribution of anionic lipids alters the structure and interaction with lipids of an amyloidogenic protein dimer bound to the membrane surface
Abstract
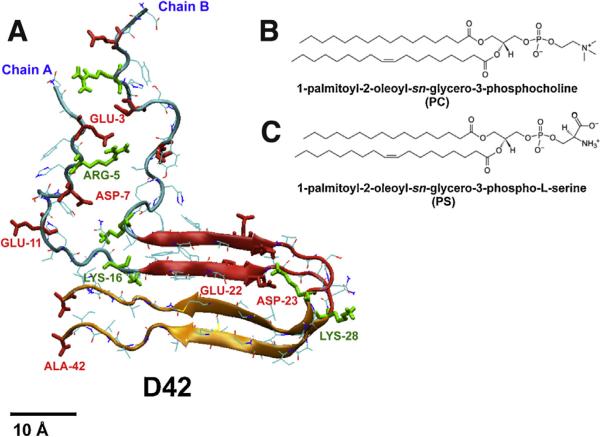
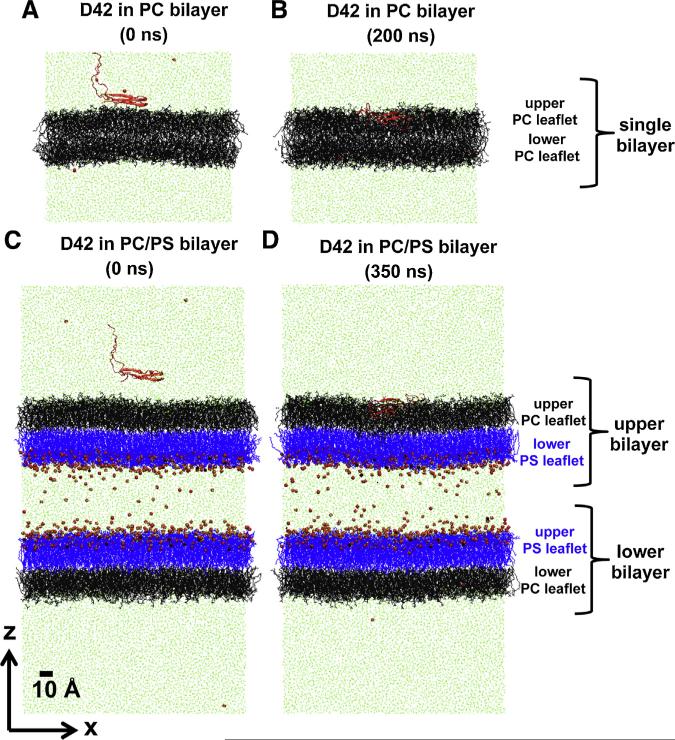
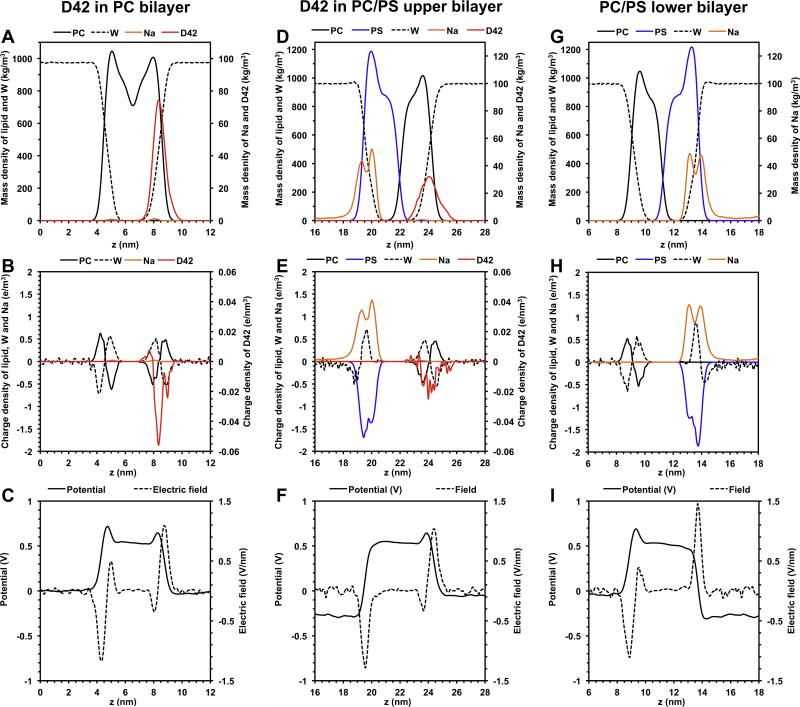
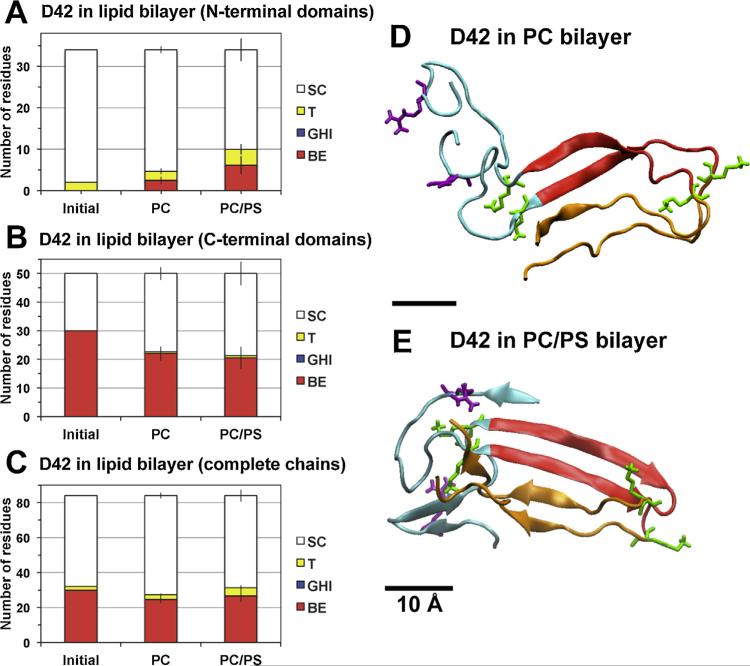
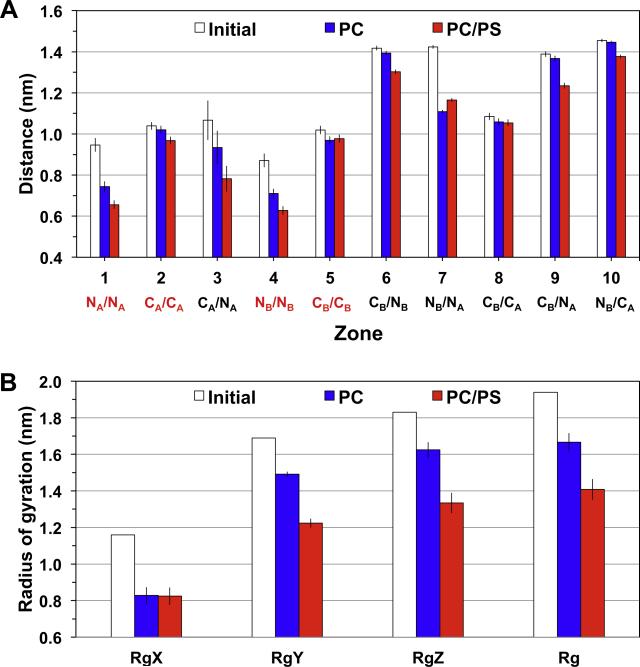
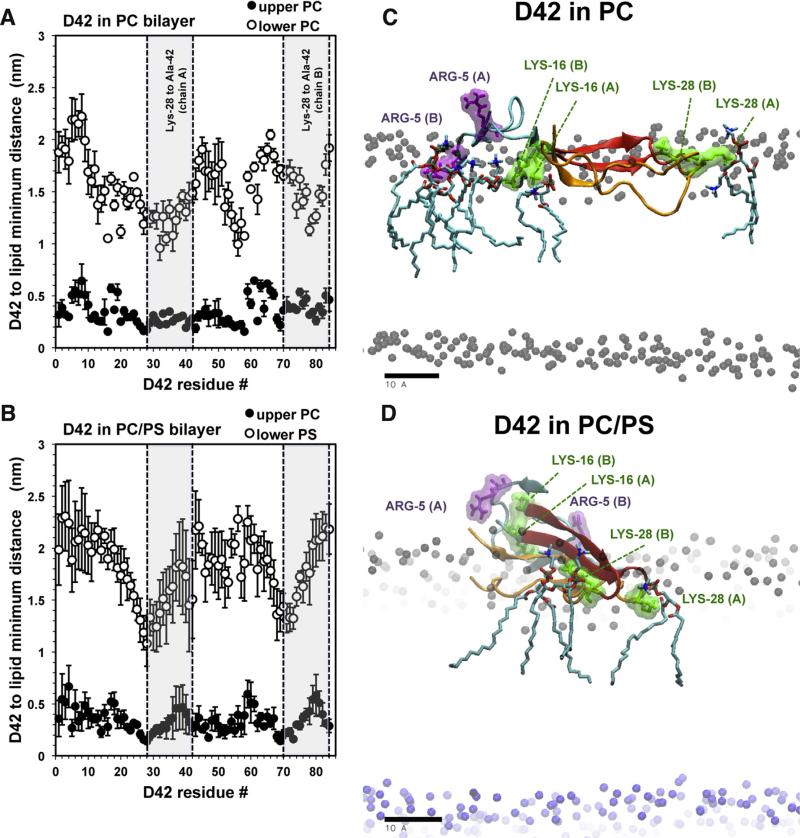
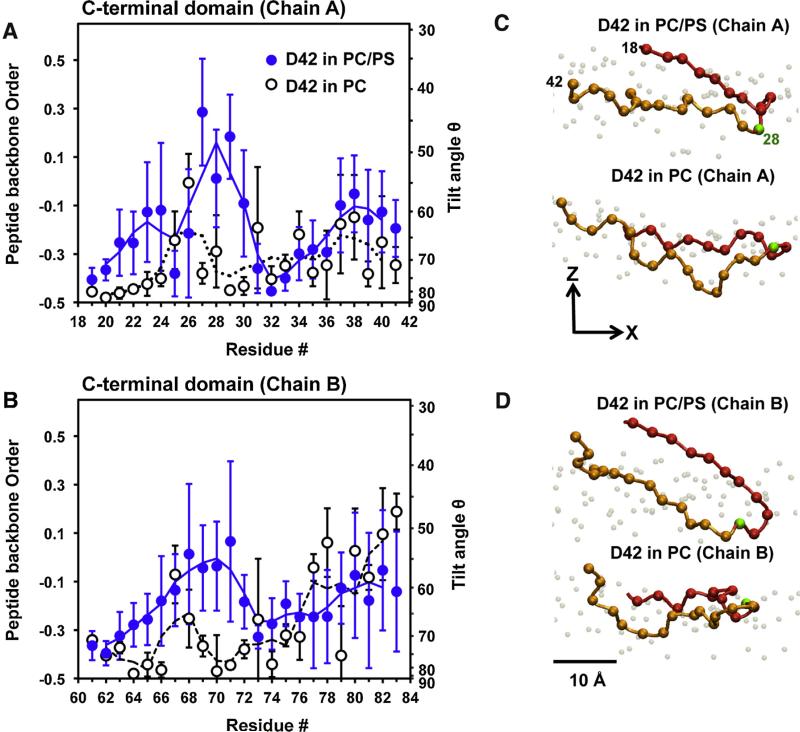
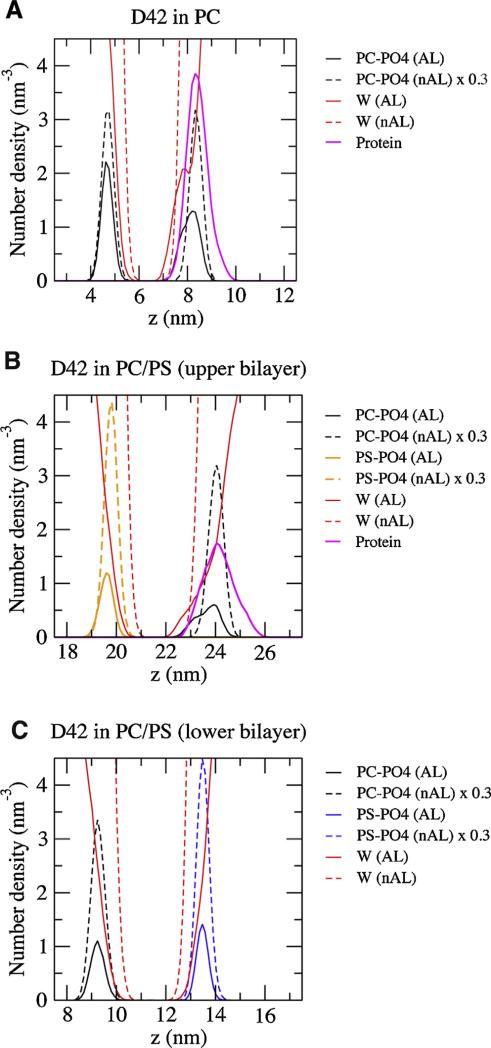
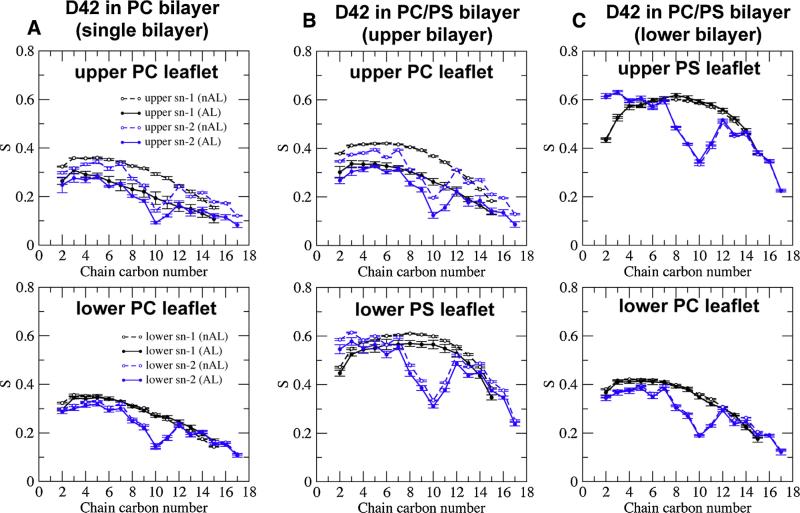
We used molecular dynamics simulations to explore the effects of asymmetric transbilayer distribution of anionic phosphatidylserine (PS) lipids on the structure of a protein on the membrane surface and subsequent protein-lipid interactions. Our simulation systems consisted of an amyloidogenic, beta-sheet rich dimeric protein (D42) absorbed to the phosphatidylcholine (PC) leaflet, or protein-contact PC leaflet, of two membrane systems: a single-component PC bilayer and double PC/PS bilayers. The latter comprised of a stable but asymmetric transbilayer distribution of PS in the presence of counterions, with a 1-component PC leaflet coupled to a 1-component PS leaflet in each bilayer. The maximally asymmetric PC/PS bilayer had a non-zero transmembrane potential (TMP) difference and higher lipid order packing, whereas the symmetric PC bilayer had a zero TMP difference and lower lipid order packing under physiologically relevant conditions. Analysis of the adsorbed protein structures revealed weaker protein binding, more folding in the N-terminal domain, more aggregation of the N- and C-terminal domains and larger tilt angle of D42 on the PC leaflet surface of the PC/PS bilayer versus the PC bilayer. Also, analysis of protein-induced membrane structural disruption revealed more localized bilayer thinning in the PC/PS versus PC bilayer. Although the electric field profile in the non-protein-contact PS leaflet of the PC/PS bilayer differed significantly from that in the non-protein-contact PC leaflet of the PC bilayer, no significant difference in the electric field profile in the protein-contact PC leaflet of either bilayer was evident. We speculate that lipid packing has a larger effect on the surface adsorbed protein structure than the electric field for a maximally asymmetric PC/PS bilayer. Our results support the mechanism that the higher lipid packing in a lipid leaflet promotes stronger protein-protein but weaker protein-lipid interactions for a dimeric protein on membrane surfaces.
Keywords: Asymmetric lipid membrane; Beta-amyloid; Molecular dynamics simulations; Protein aggregation; Protein structures on surfaces; Protein–lipid interactions.
Copyright © 2016 Elsevier Ireland Ltd. All rights reserved.
Figures

Similar articles
-
Data supporting beta-amyloid dimer structural transitions and protein-lipid interactions on asymmetric lipid bilayer surfaces using MD simulations on experimentally derived NMR protein structures.Data Brief. 2016 Mar 10;7:658-72. doi: 10.1016/j.dib.2016.03.015. eCollection 2016 Jun. Data Brief. 2016. PMID: 27054174 Free PMC article.
-
Dual Role of Anionic Lipids in Amyloid Aggregation.J Phys Chem B. 2024 Nov 7;128(44):10831-10840. doi: 10.1021/acs.jpcb.4c05636. Epub 2024 Oct 25. J Phys Chem B. 2024. PMID: 39450869
-
Mechanical properties of anionic asymmetric bilayers from atomistic simulations.J Chem Phys. 2021 Jun 14;154(22):224701. doi: 10.1063/5.0048232. J Chem Phys. 2021. PMID: 34241213 Free PMC article.
-
Asymmetric distribution of phospholipids in biomembranes.Biol Pharm Bull. 2006 Aug;29(8):1547-53. doi: 10.1248/bpb.29.1547. Biol Pharm Bull. 2006. PMID: 16880602 Review.
-
Polyelectrolyte-induced domains in lipid bilayer membranes: the deuterium NMR perspective.Biochem Cell Biol. 1998;76(2-3):452-64. doi: 10.1139/bcb-76-2-3-452. Biochem Cell Biol. 1998. PMID: 9923714 Review.
Cited by
-
Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation.Chem Rev. 2019 May 8;119(9):6086-6161. doi: 10.1021/acs.chemrev.8b00608. Epub 2019 Apr 12. Chem Rev. 2019. PMID: 30978005 Free PMC article. Review.
-
Data supporting beta-amyloid dimer structural transitions and protein-lipid interactions on asymmetric lipid bilayer surfaces using MD simulations on experimentally derived NMR protein structures.Data Brief. 2016 Mar 10;7:658-72. doi: 10.1016/j.dib.2016.03.015. eCollection 2016 Jun. Data Brief. 2016. PMID: 27054174 Free PMC article.
-
Exploring Membrane Binding Targets of Disordered Human Tau Aggregates on Lipid Rafts Using Multiscale Molecular Dynamics Simulations.Membranes (Basel). 2022 Nov 4;12(11):1098. doi: 10.3390/membranes12111098. Membranes (Basel). 2022. PMID: 36363654 Free PMC article.
-
Coarse-grained MD simulations reveal beta-amyloid fibrils of various sizes bind to interfacial liquid-ordered and liquid-disordered regions in phase separated lipid rafts with diverse membrane-bound conformational states.Biophys Chem. 2020 May;260:106355. doi: 10.1016/j.bpc.2020.106355. Epub 2020 Mar 5. Biophys Chem. 2020. PMID: 32179374 Free PMC article.
-
Molecular insights into the primary nucleation of polymorphic amyloid β dimers in DOPC lipid bilayer membrane.Protein Sci. 2022 May;31(5):e4283. doi: 10.1002/pro.4283. Protein Sci. 2022. PMID: 35129859 Free PMC article.
References
-
- Allen WJ, Lemkul JA, Bevan DR. GridMAT-MD: a grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009;30:1952–1958. - PubMed
-
- Basyn F, Charloteaux B, Thomas A, Brasseur R. Prediction of membrane protein orientation in lipid bilayers: a theoretical approach. J. Mol. Graph Model. 2001;20:235–244. - PubMed
-
- Bekker H, Berendsen HJC, Dijkstra EJ, Achterop S, van Drunen R, van der Spoel D, Sijbers A, Keegstra H, Reitsma B, Renardus MKR. Physics Computing 92. World Scientific; Singapore: 1993. Gromacs: A Parallel Computer for Molecular Dynamics Simulations.
-
- Berendsen HD, van der Spoel D, van Drunen R. GROMACS: a messagepassing parallel molecular dynamics implementation. Comp. Phys. Comm. 1995;91:43–56.
-
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
