

Skin Microbiome Surveys Are Strongly Influenced by Experimental Design
- PMID: 26829039
- PMCID: PMC4842136
- DOI: 10.1016/j.jid.2016.01.016
Skin Microbiome Surveys Are Strongly Influenced by Experimental Design
Abstract
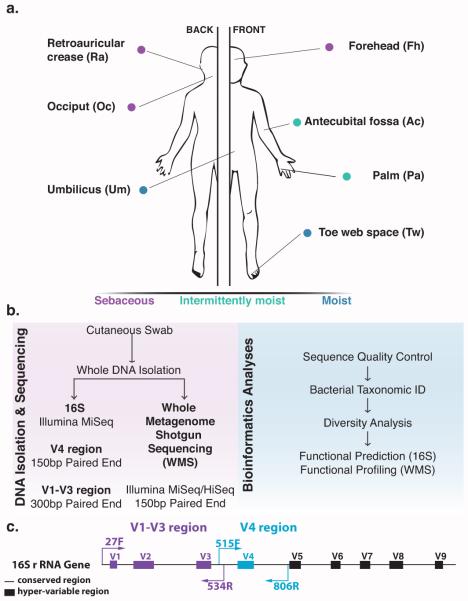
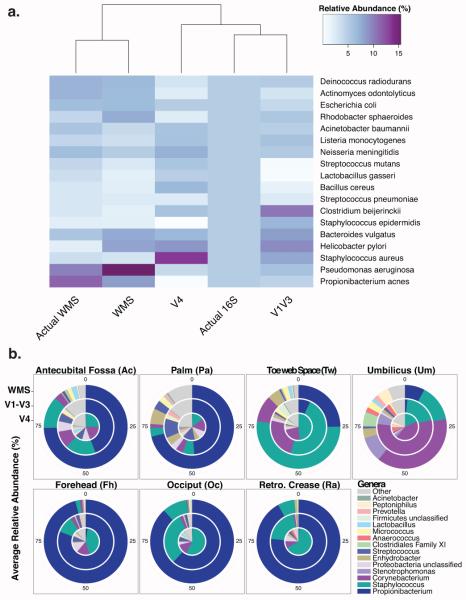
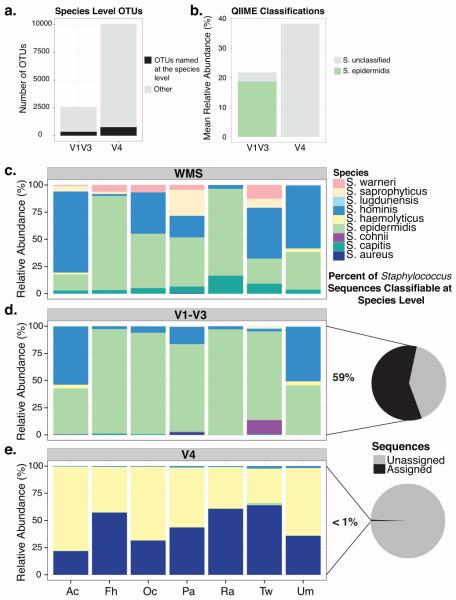
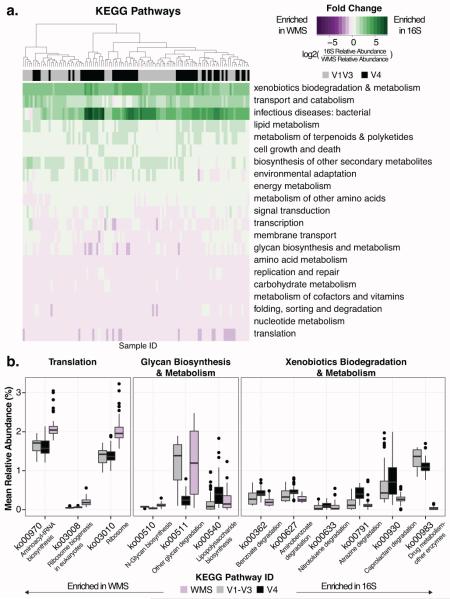
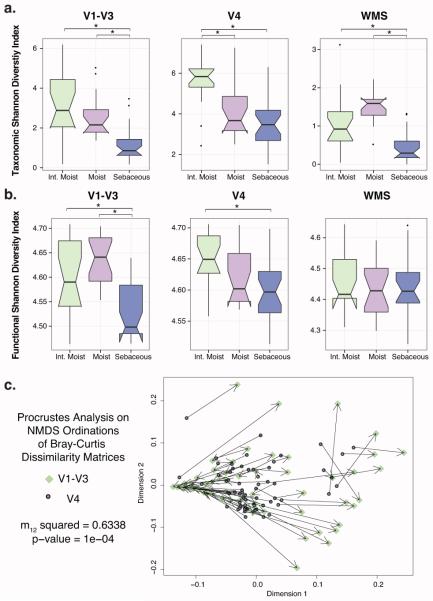
Culture-independent studies to characterize skin microbiota are increasingly common, due in part to affordable and accessible sequencing and analysis platforms. Compared to culture-based techniques, DNA sequencing of the bacterial 16S ribosomal RNA (rRNA) gene or whole metagenome shotgun (WMS) sequencing provides more precise microbial community characterizations. Most widely used protocols were developed to characterize microbiota of other habitats (i.e., gastrointestinal) and have not been systematically compared for their utility in skin microbiome surveys. Here we establish a resource for the cutaneous research community to guide experimental design in characterizing skin microbiota. We compare two widely sequenced regions of the 16S rRNA gene to WMS sequencing for recapitulating skin microbiome community composition, diversity, and genetic functional enrichment. We show that WMS sequencing most accurately recapitulates microbial communities, but sequencing of hypervariable regions 1-3 of the 16S rRNA gene provides highly similar results. Sequencing of hypervariable region 4 poorly captures skin commensal microbiota, especially Propionibacterium. WMS sequencing, which is resource and cost intensive, provides evidence of a community's functional potential; however, metagenome predictions based on 16S rRNA sequence tags closely approximate WMS genetic functional profiles. This study highlights the importance of experimental design for downstream results in skin microbiome surveys.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
Reply to Meisel et al.J Invest Dermatol. 2017 Apr;137(4):961-962. doi: 10.1016/j.jid.2016.11.013. Epub 2016 Nov 22. J Invest Dermatol. 2017. PMID: 27887953 No abstract available.
References
-
- Albertsen M, Karst SM, Ziegler AS, Kirkegaard RH, Nielsen PH. Aziz RK, editor. Back to Basics – The Influence of DNA Extraction and Primer Choice on Phylogenetic Analysis of Activated Sludge Communities. PloS one [Internet] 2015 Jul 16;10(7):e0132783–15. Available from: http://dx.plos.org/10.1371/journal.pone.0132783. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
