

An Annotation Agnostic Algorithm for Detecting Nascent RNA Transcripts in GRO-Seq
- PMID: 26829802
- PMCID: PMC5667649
- DOI: 10.1109/TCBB.2016.2520919
An Annotation Agnostic Algorithm for Detecting Nascent RNA Transcripts in GRO-Seq
Abstract
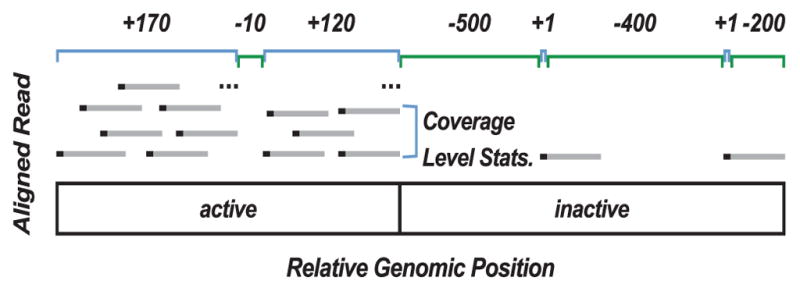
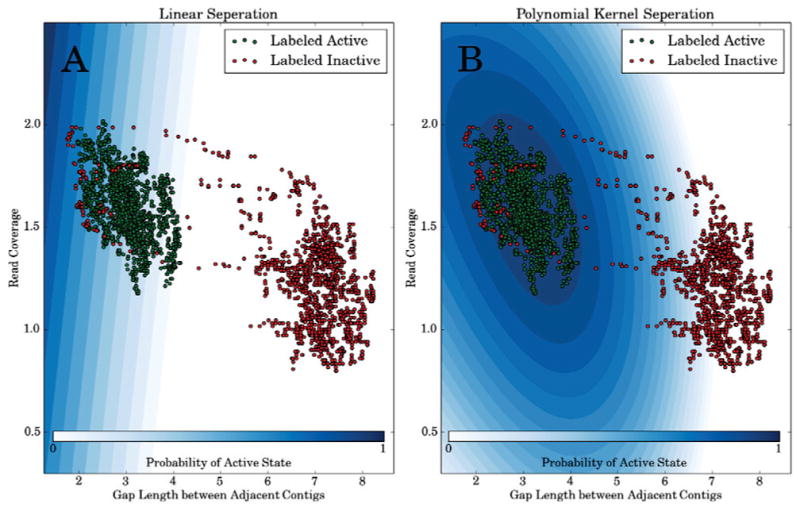
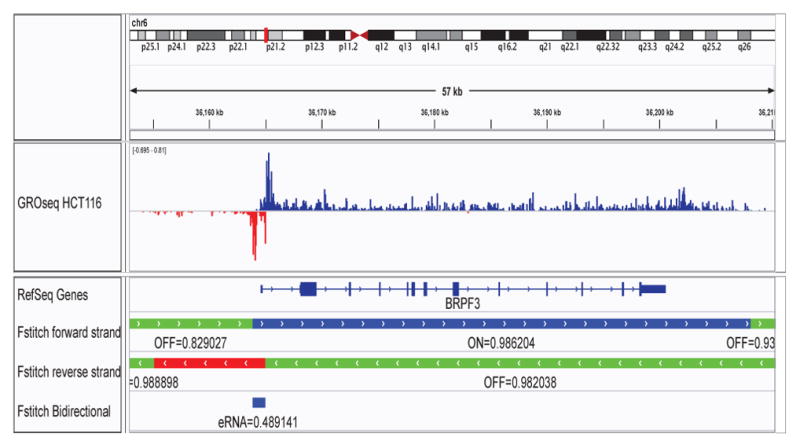
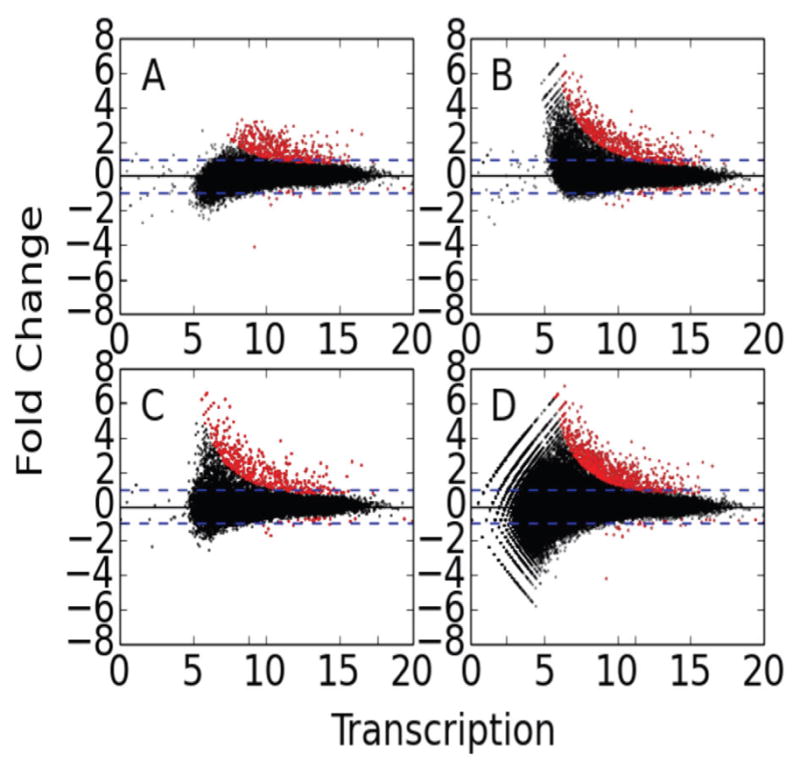
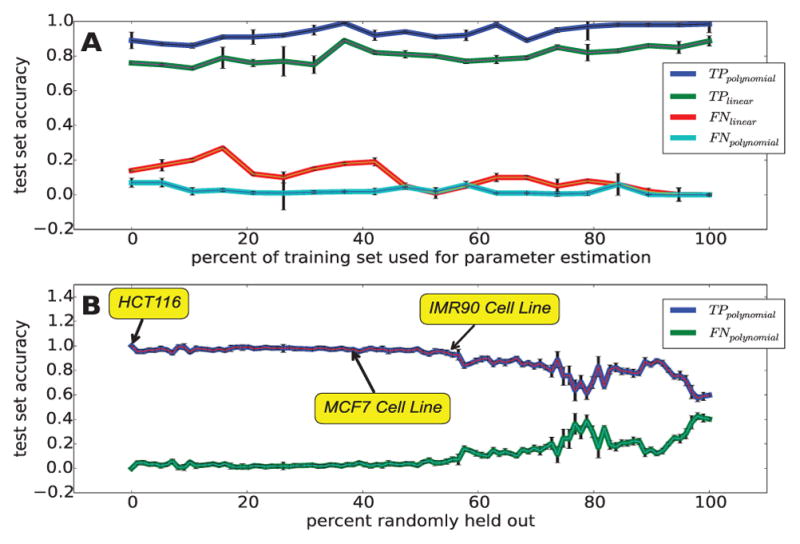
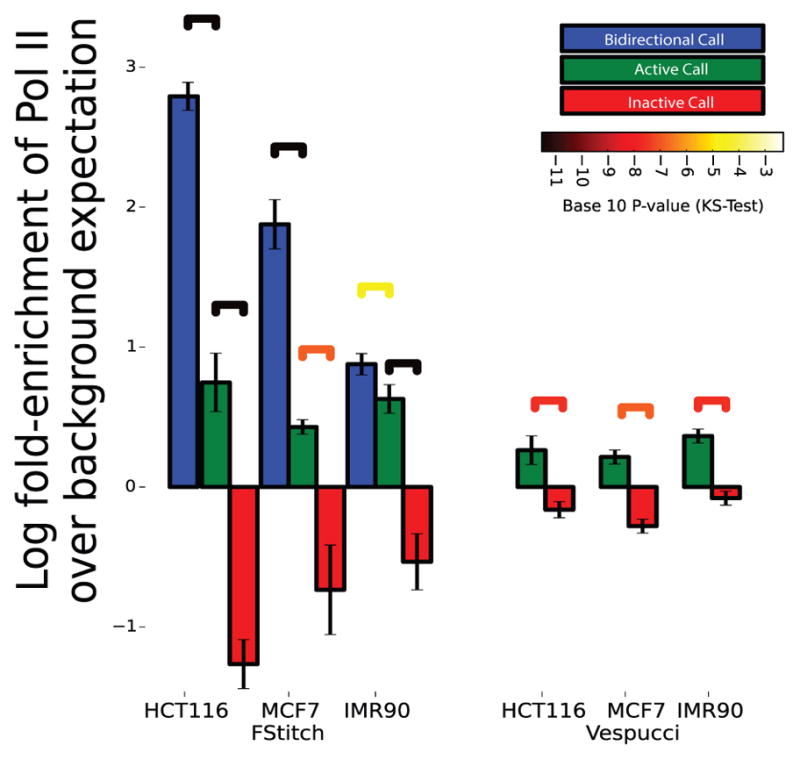
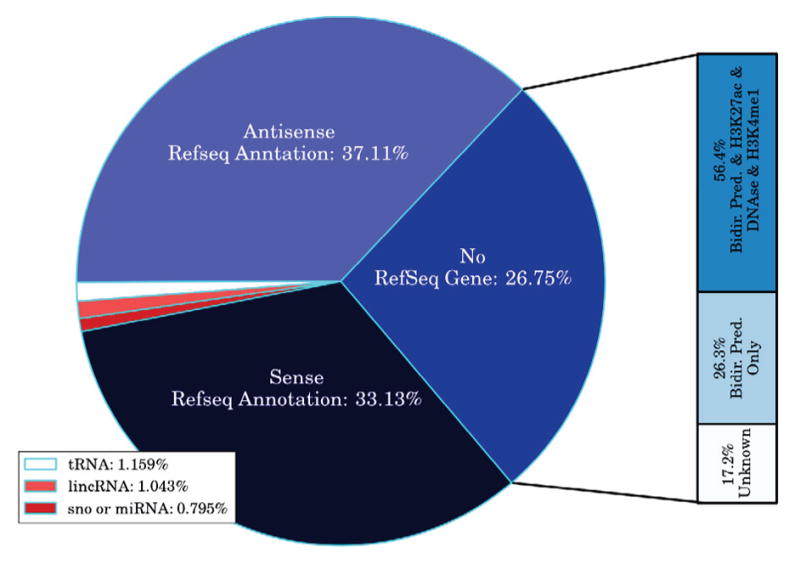
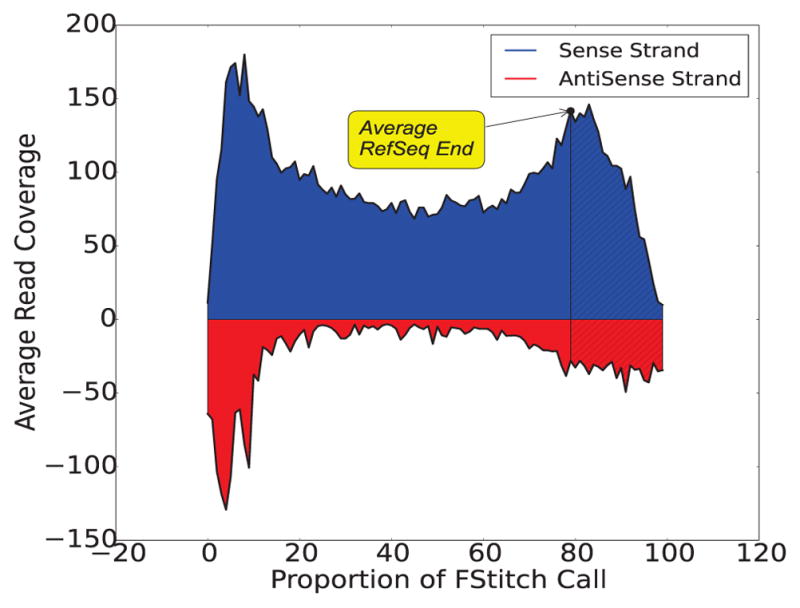
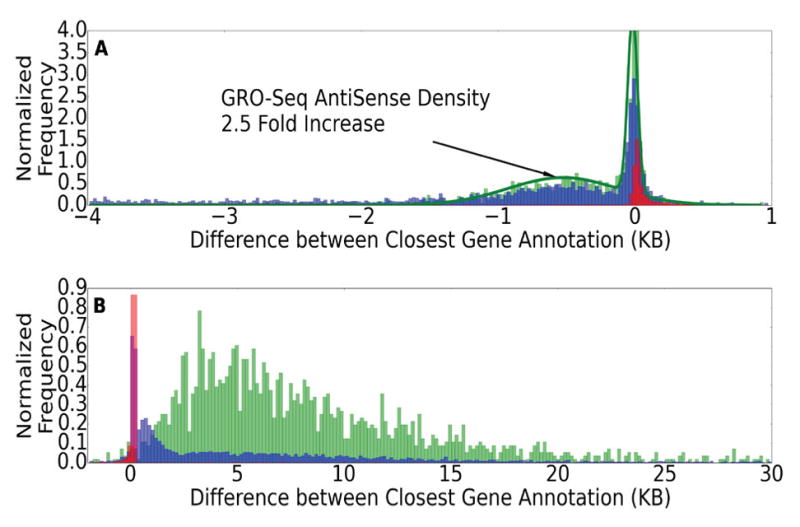
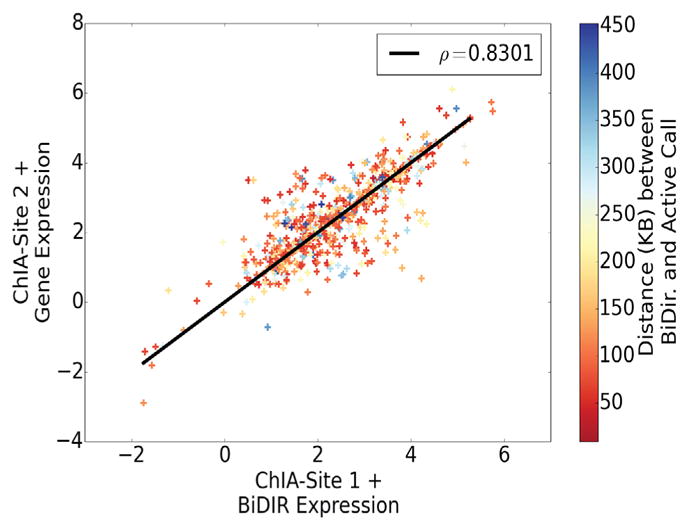
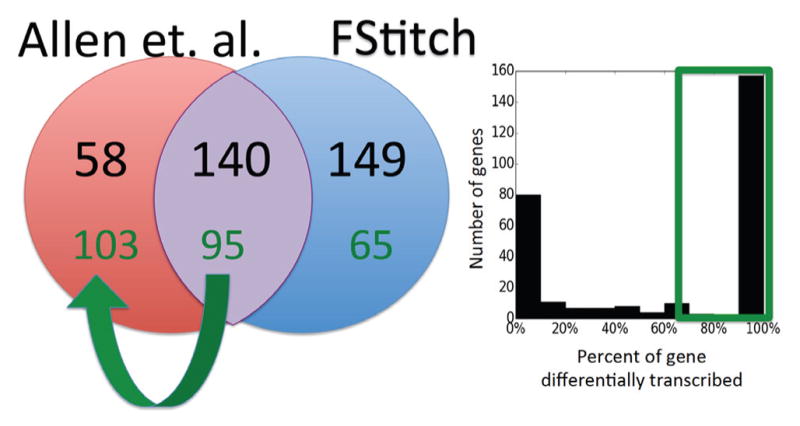
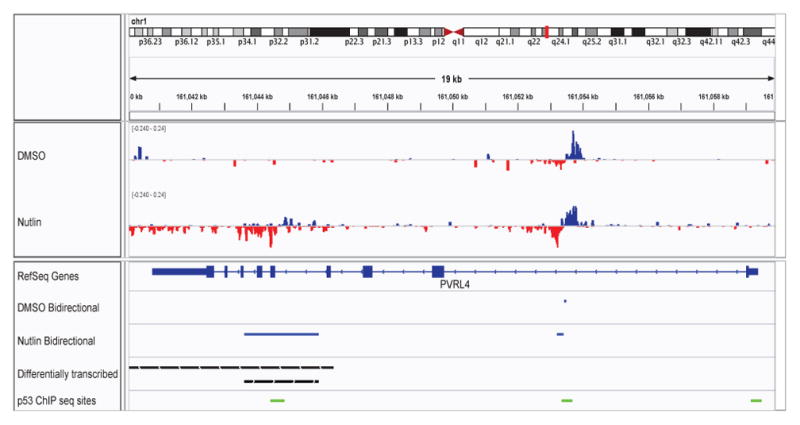
We present a fast and simple algorithm to detect nascent RNA transcription in global nuclear run-on sequencing (GRO-seq). GRO-seq is a relatively new protocol that captures nascent transcripts from actively engaged polymerase, providing a direct read-out on bona fide transcription. Most traditional assays, such as RNA-seq, measure steady state RNA levels which are affected by transcription, post-transcriptional processing, and RNA stability. GRO-seq data, however, presents unique analysis challenges that are only beginning to be addressed. Here, we describe a new algorithm, Fast Read Stitcher (FStitch), that takes advantage of two popular machine-learning techniques, hidden Markov models and logistic regression, to classify which regions of the genome are transcribed. Given a small user-defined training set, our algorithm is accurate, robust to varying read depth, annotation agnostic, and fast. Analysis of GRO-seq data without a priori need for annotation uncovers surprising new insights into several aspects of the transcription process.
Figures
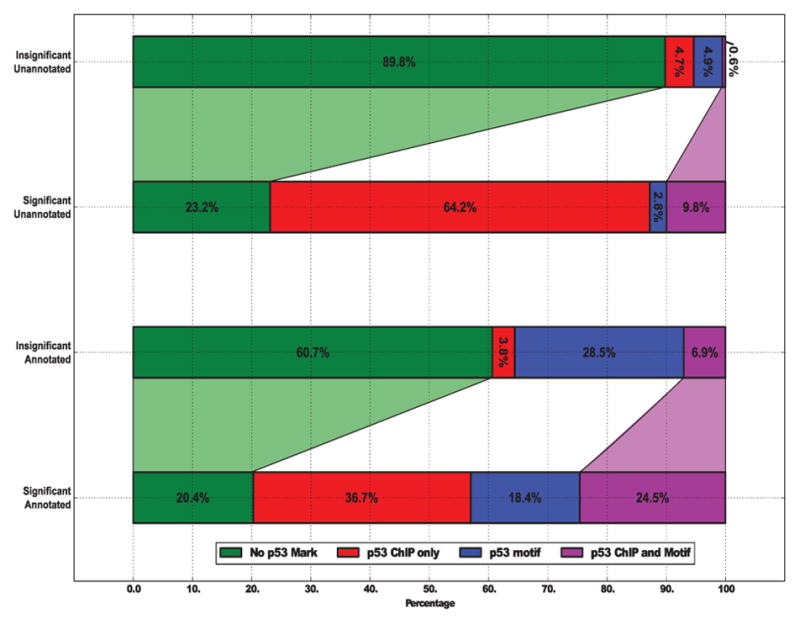
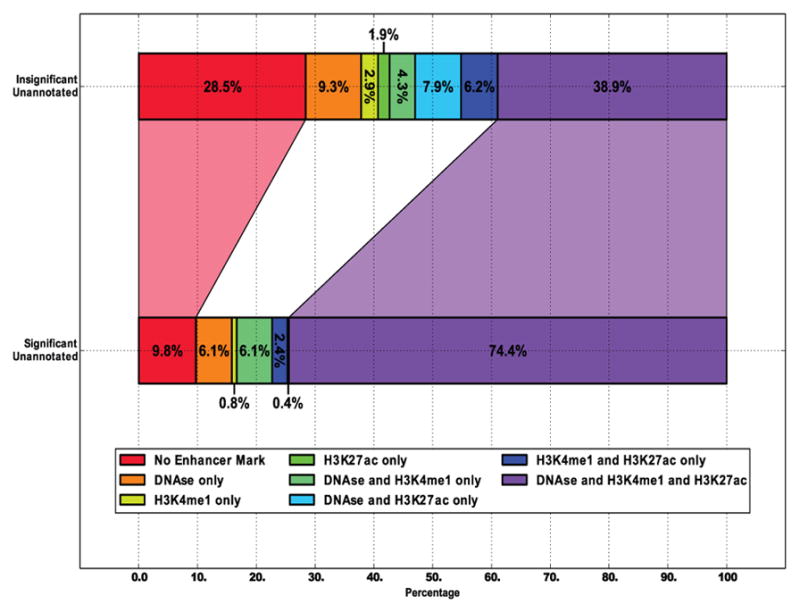
References
-
- Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8(6):413–423. - PubMed
-
- Min I, Waterfall J, Core L, Munroe R, Schimenti J, Lis J. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes & Development. 2011;25(7):742–754. [Online]. Available: http://genesdev.cshlp.org/content/25/7/742.abstract. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
