

Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits
- PMID: 26830879
- PMCID: PMC4752902
- DOI: 10.1016/j.cell.2016.01.011
Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits
Abstract
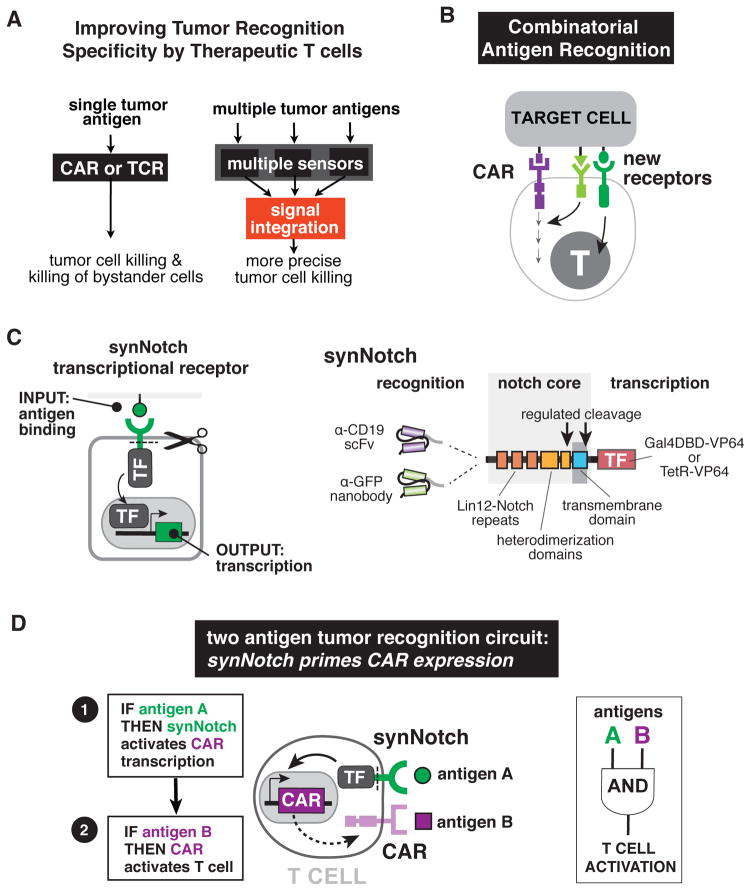
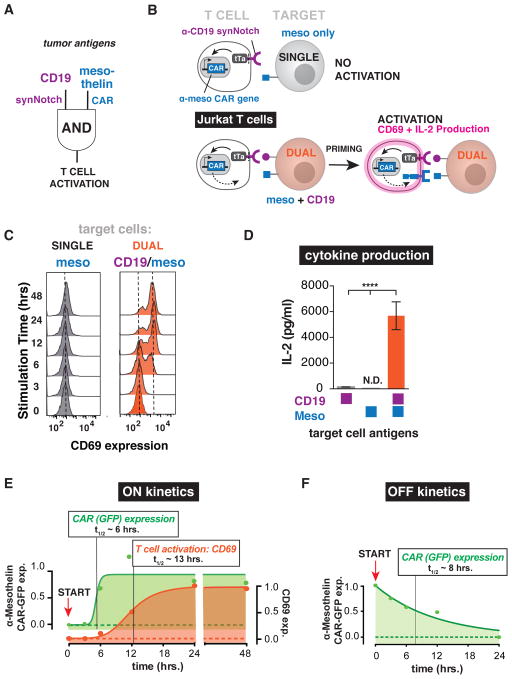
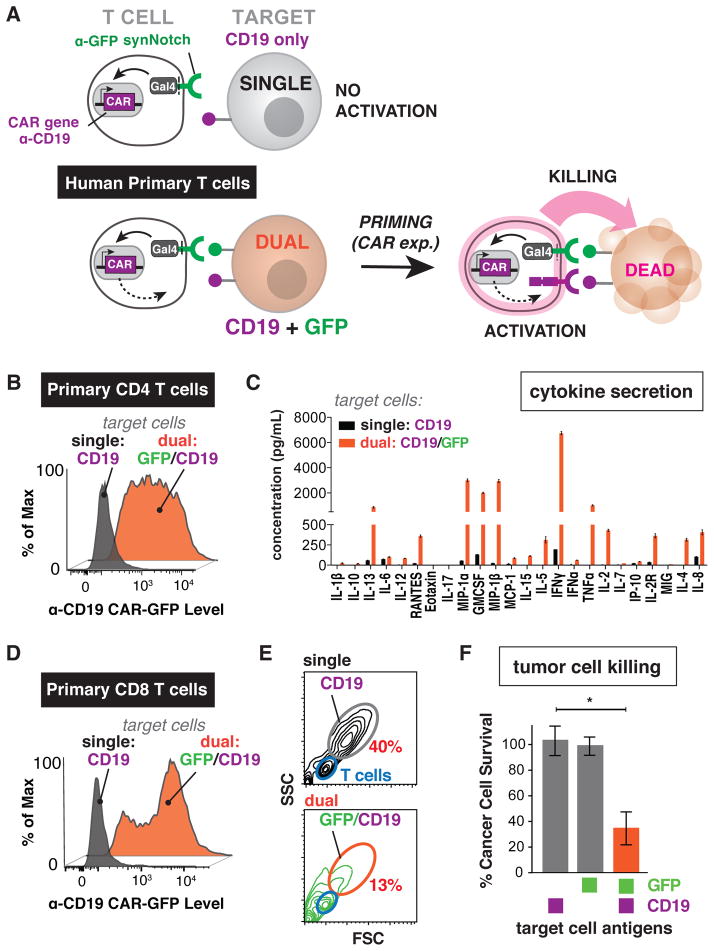
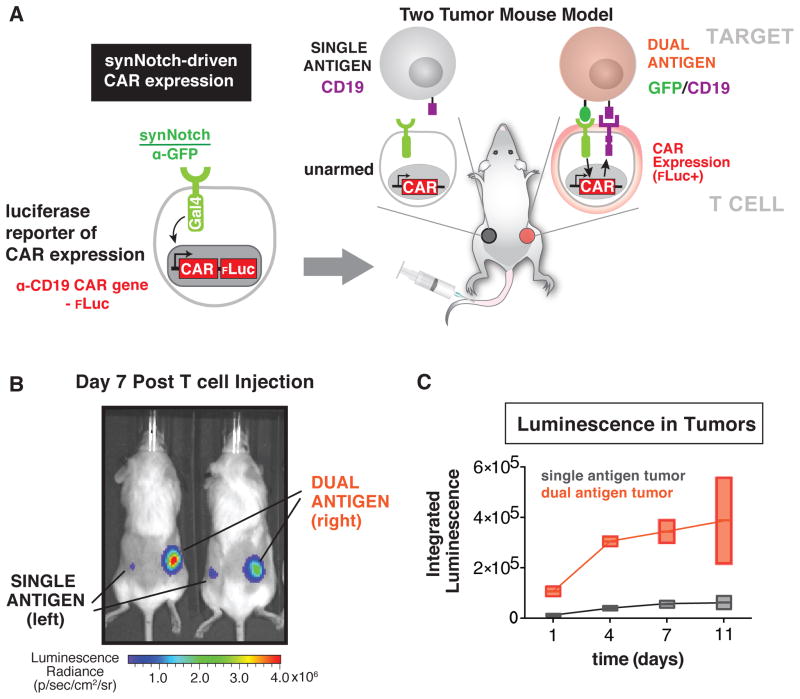
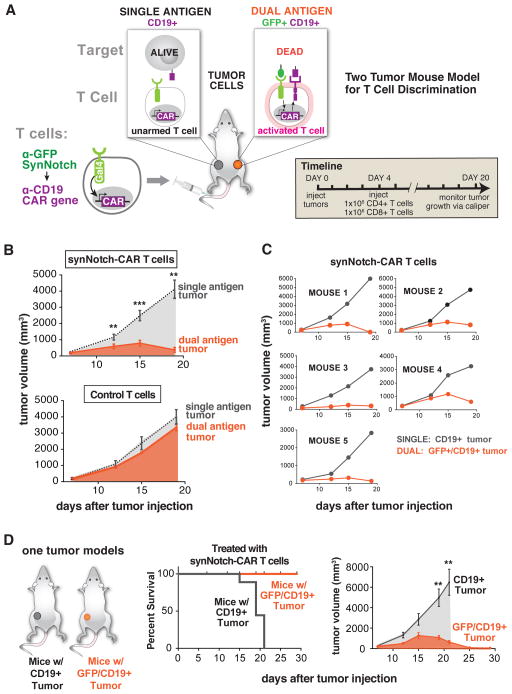
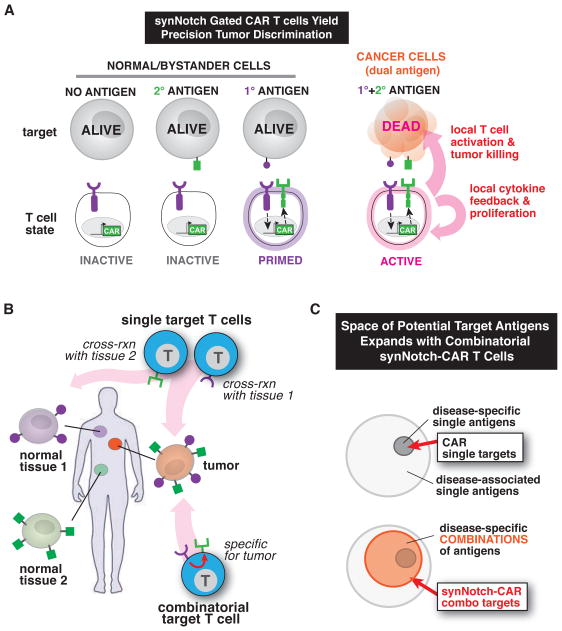
T cells can be re-directed to kill cancer cells using chimeric antigen receptors (CARs) or T cell receptors (TCRs). This approach, however, is constrained by the rarity of tumor-specific single antigens. Targeting antigens also found on bystander tissues can cause life-threatening adverse effects. A powerful way to enhance ON-target activity of therapeutic T cells is to engineer them to require combinatorial antigens. Here, we engineer a combinatorially activated T cell circuit in which a synthetic Notch receptor for one antigen induces the expression of a CAR for a second antigen. These dual-receptor AND-gate T cells are only armed and activated in the presence of dual antigen tumor cells. These T cells show precise therapeutic discrimination in vivo-sparing single antigen "bystander" tumors while efficiently clearing combinatorial antigen "disease" tumors. This type of precision dual-receptor circuit opens the door to immune recognition of a wider range of tumors. VIDEO ABSTRACT.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Comment in
-
A Receptor for All Occasions.Cell. 2016 Feb 11;164(4):599-600. doi: 10.1016/j.cell.2016.01.030. Cell. 2016. PMID: 26871625
-
Immunotherapy: Two antigens are better than one.Nat Rev Cancer. 2016 Mar;16(3):128-9. doi: 10.1038/nrc.2016.17. Epub 2016 Feb 19. Nat Rev Cancer. 2016. PMID: 26893068 No abstract available.
-
Combinatorial Antigen Targeting: Ideal T-Cell Sensing and Anti-Tumor Response.Trends Mol Med. 2016 Apr;22(4):271-273. doi: 10.1016/j.molmed.2016.02.009. Epub 2016 Mar 10. Trends Mol Med. 2016. PMID: 26971630 Free PMC article.
-
Receptor combinations hone T-cell therapy.Nat Biotechnol. 2016 Apr;34(4):389-91. doi: 10.1038/nbt.3539. Nat Biotechnol. 2016. PMID: 27054992 No abstract available.
-
SYNTHETIC BIOLOGY. Customizing cell-cell communication.Nat Methods. 2016 Apr;13(4):285. doi: 10.1038/nmeth.3824. Nat Methods. 2016. PMID: 27482567 No abstract available.
