

α1A-Adrenergic receptor prevents cardiac ischemic damage through PKCδ/GLUT1/4-mediated glucose uptake
- PMID: 26832303
- PMCID: PMC4740921
- DOI: 10.3109/10799893.2015.1091475
α1A-Adrenergic receptor prevents cardiac ischemic damage through PKCδ/GLUT1/4-mediated glucose uptake
Abstract
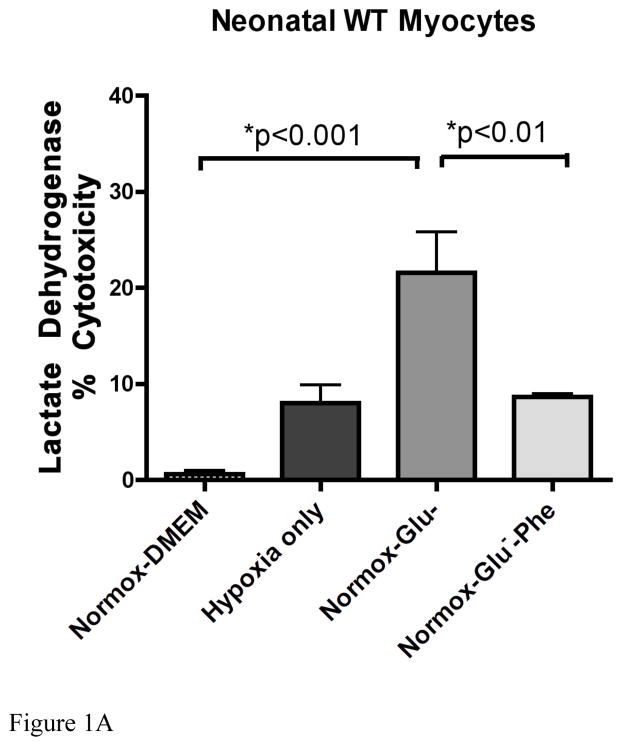
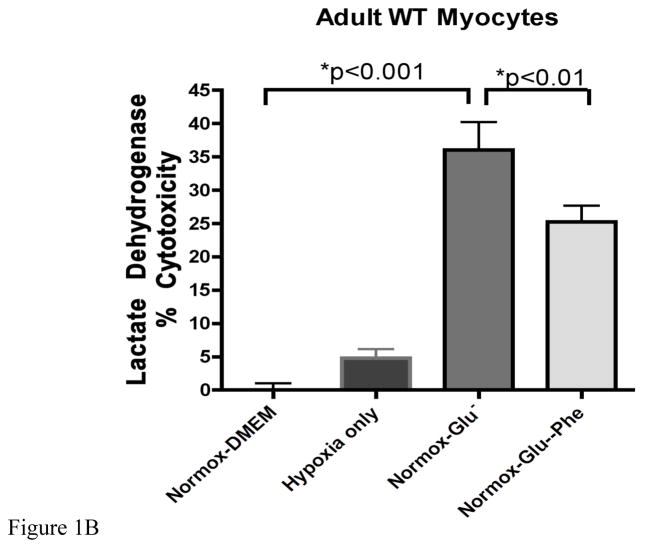
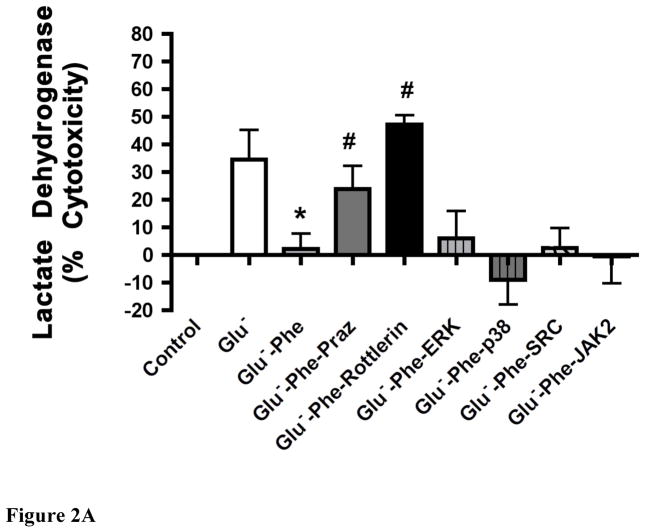
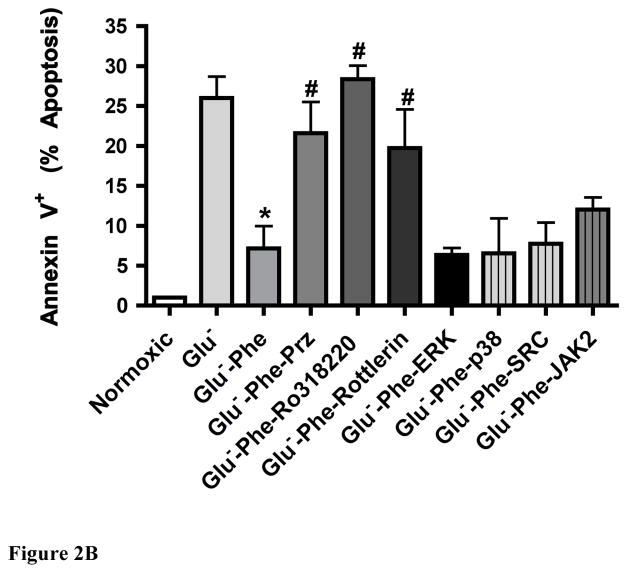
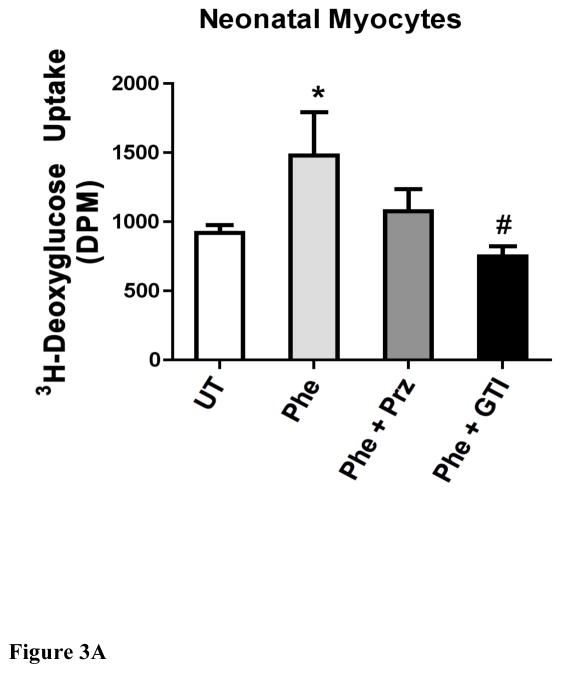
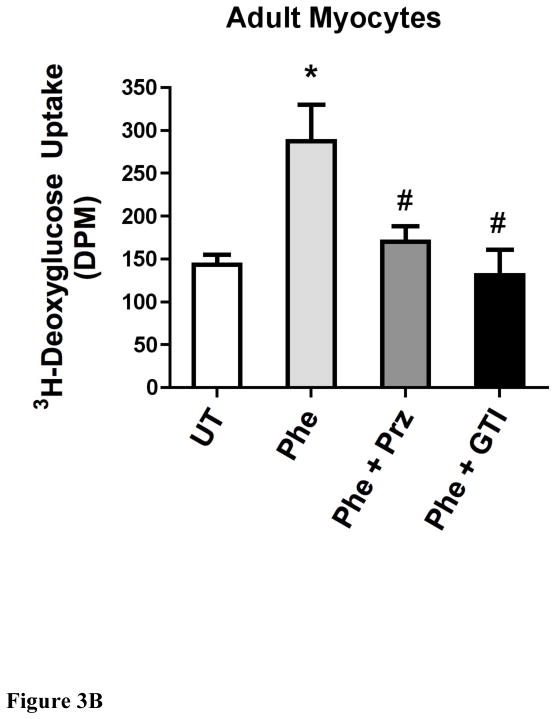
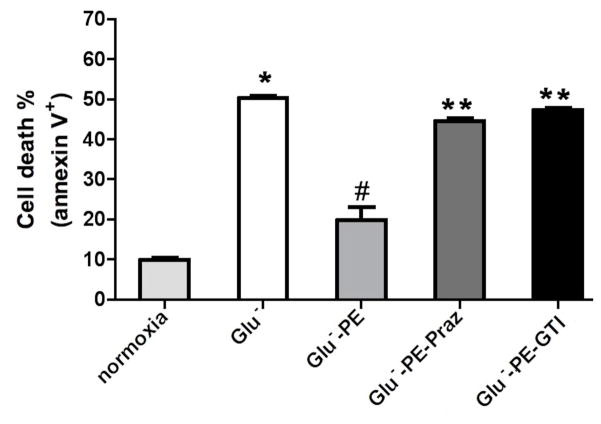
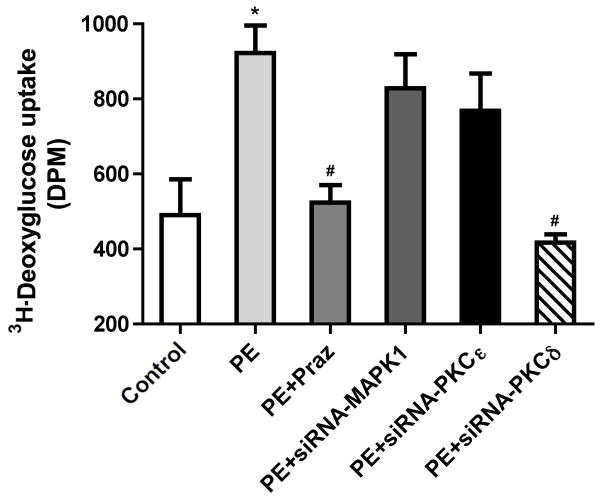
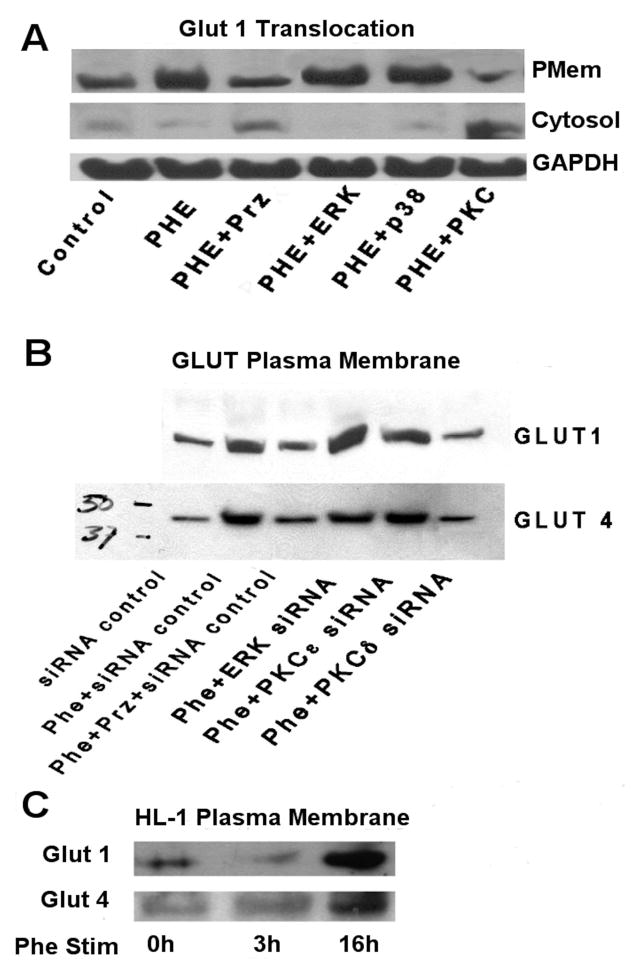
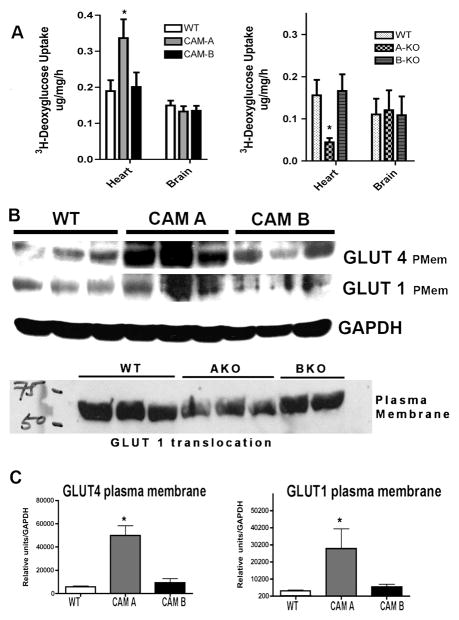
While α(1)-adrenergic receptors (ARs) have been previously shown to limit ischemic cardiac damage, the mechanisms remain unclear. Most previous studies utilized low oxygen conditions in addition to ischemic buffers with glucose deficiencies, but we discovered profound differences if the two conditions are separated. We assessed both mouse neonatal and adult myocytes and HL-1 cells in a series of assays assessing ischemic damage under hypoxic or low glucose conditions. We found that α(1)-AR stimulation protected against increased lactate dehydrogenase release or Annexin V(+) apoptosis under conditions that were due to low glucose concentration not to hypoxia. The α(1)-AR antagonist prazosin or nonselective protein kinase C (PKC) inhibitors blocked the protective effect. α(1)-AR stimulation increased (3)H-deoxyglucose uptake that was blocked with either an inhibitor to glucose transporter 1 or 4 (GLUT1 or GLUT4) or small interfering RNA (siRNA) against PKCδ. GLUT1/4 inhibition also blocked α(1)-AR-mediated protection from apoptosis. The PKC inhibitor rottlerin or siRNA against PKCδ blocked α(1)-AR stimulated GLUT1 or GLUT4 plasma membrane translocation. α(1)-AR stimulation increased plasma membrane concentration of either GLUT1 or GLUT4 in a time-dependent fashion. Transgenic mice overexpressing the α(1A)-AR but not α(1B)-AR mice displayed increased glucose uptake and increased GLUT1 and GLUT4 plasma membrane translocation in the adult heart while α(1A)-AR but not α(1B)-AR knockout mice displayed lowered glucose uptake and GLUT translocation. Our results suggest that α(1)-AR activation is anti-apoptotic and protective during cardiac ischemia due to glucose deprivation and not hypoxia by enhancing glucose uptake into the heart via PKCδ-mediated GLUT translocation that may be specific to the α(1A)-AR subtype.
Keywords: Adrenergic receptor; cardiac; glucose transporter.
Conflict of interest statement
Figures
Similar articles
-
Contribution of alpha-adrenergic and beta-adrenergic stimulation to ischemia-induced glucose transporter (GLUT) 4 and GLUT1 translocation in the isolated perfused rat heart.Circ Res. 1999 Jun 25;84(12):1407-15. doi: 10.1161/01.res.84.12.1407. Circ Res. 1999. PMID: 10381893
-
Beta3-adrenergic receptors stimulate glucose uptake in brown adipocytes by two mechanisms independently of glucose transporter 4 translocation.Endocrinology. 2006 Dec;147(12):5730-9. doi: 10.1210/en.2006-0242. Epub 2006 Sep 7. Endocrinology. 2006. PMID: 16959848
-
α(1A)-adrenergic receptor differentially regulates STAT3 phosphorylation through PKCϵ and PKCδ in myocytes.J Recept Signal Transduct Res. 2012 Apr;32(2):76-86. doi: 10.3109/10799893.2011.647353. Epub 2012 Jan 24. J Recept Signal Transduct Res. 2012. PMID: 22268811 Free PMC article.
-
Regulation of glucose transport by hypoxia.Am J Kidney Dis. 1999 Jul;34(1):189-202. doi: 10.1016/s0272-6386(99)70131-9. Am J Kidney Dis. 1999. PMID: 10401038 Review.
-
Regulation of myocardial glucose uptake and transport during ischemia and energetic stress.Am J Cardiol. 1999 Jun 17;83(12A):25H-30H. doi: 10.1016/s0002-9149(99)00253-2. Am J Cardiol. 1999. PMID: 10750583 Review.
Cited by
-
Characterization of a novel positive allosteric modulator of the α1A-Adrenergic receptor.Curr Res Pharmacol Drug Discov. 2022 Dec 2;4:100142. doi: 10.1016/j.crphar.2022.100142. eCollection 2023. Curr Res Pharmacol Drug Discov. 2022. PMID: 36544813 Free PMC article.
-
Enhancement of Glucose Metabolism via PGC-1α Participates in the Cardioprotection of Chronic Intermittent Hypobaric Hypoxia.Front Physiol. 2016 Jun 8;7:219. doi: 10.3389/fphys.2016.00219. eCollection 2016. Front Physiol. 2016. PMID: 27375497 Free PMC article.
-
Novel Structural Approaches to Study GPCR Regulation.Int J Mol Sci. 2016 Dec 23;18(1):27. doi: 10.3390/ijms18010027. Int J Mol Sci. 2016. PMID: 28025563 Free PMC article. Review.
-
A comparison of the metabolic side-effects of the second-generation antipsychotic drugs risperidone and paliperidone in animal models.PLoS One. 2021 Jan 28;16(1):e0246211. doi: 10.1371/journal.pone.0246211. eCollection 2021. PLoS One. 2021. PMID: 33508013 Free PMC article.
-
Changes in Myocardial Metabolism Preceding Sudden Cardiac Death.Front Physiol. 2020 Jun 16;11:640. doi: 10.3389/fphys.2020.00640. eCollection 2020. Front Physiol. 2020. PMID: 32612538 Free PMC article. Review.
References
-
- Grupp IL, Lorenz JN, Walsh RA, Boivin GP, Rindt H. Overexpression of α1B-adrenergic receptor induces left ventricular dysfunction in the absence of hypertrophy. Am J Physiol. 1998;44:H1338–H1350. - PubMed
-
- Wang BH, Du XJ, Autelitano DJ, Milano CA, Woodcock EA. Adverse effects of constitutively active α1B-adrenergic receptors after pressure overload in mouse hearts. Am J Physiol Heart Circ Physiol. 2000;279:H1079–H1086. - PubMed
-
- Zuscik MJ, Chalothorn D, Hellard D, Deighan C, McGee A, Daly CJ, Waugh DJ, Ross SA, Gaivin RJ, Morehead AJ, Thomas JD, Plow EF, McGrath JC, Piascik MT, Perez DM. Hypotension, autonomic failure, and cardiac hypertrophy in transgenic mice overexpressing the α1B-adrenergic receptor. J Biol Chem. 2001;276:13738–13743. - PubMed
-
- Lin G, Owens WA, Chen SH, Stevens ME, Kesteven S, Arthur JF, Woodcock EA, Feneley MP, Graham RM. Targeted α1A-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ Res. 2001;89:343–350. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous