

Export of malaria proteins requires co-translational processing of the PEXEL motif independent of phosphatidylinositol-3-phosphate binding
- PMID: 26832821
- PMCID: PMC4740378
- DOI: 10.1038/ncomms10470
Export of malaria proteins requires co-translational processing of the PEXEL motif independent of phosphatidylinositol-3-phosphate binding
Abstract
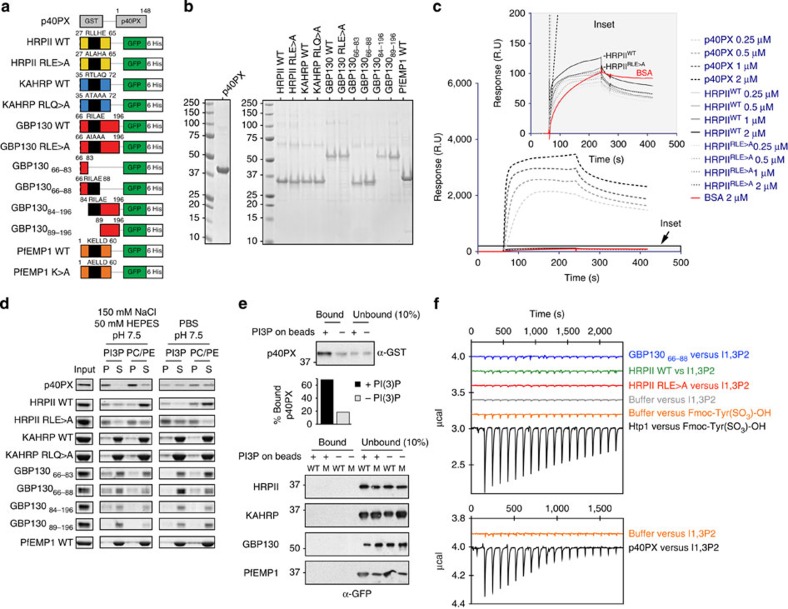
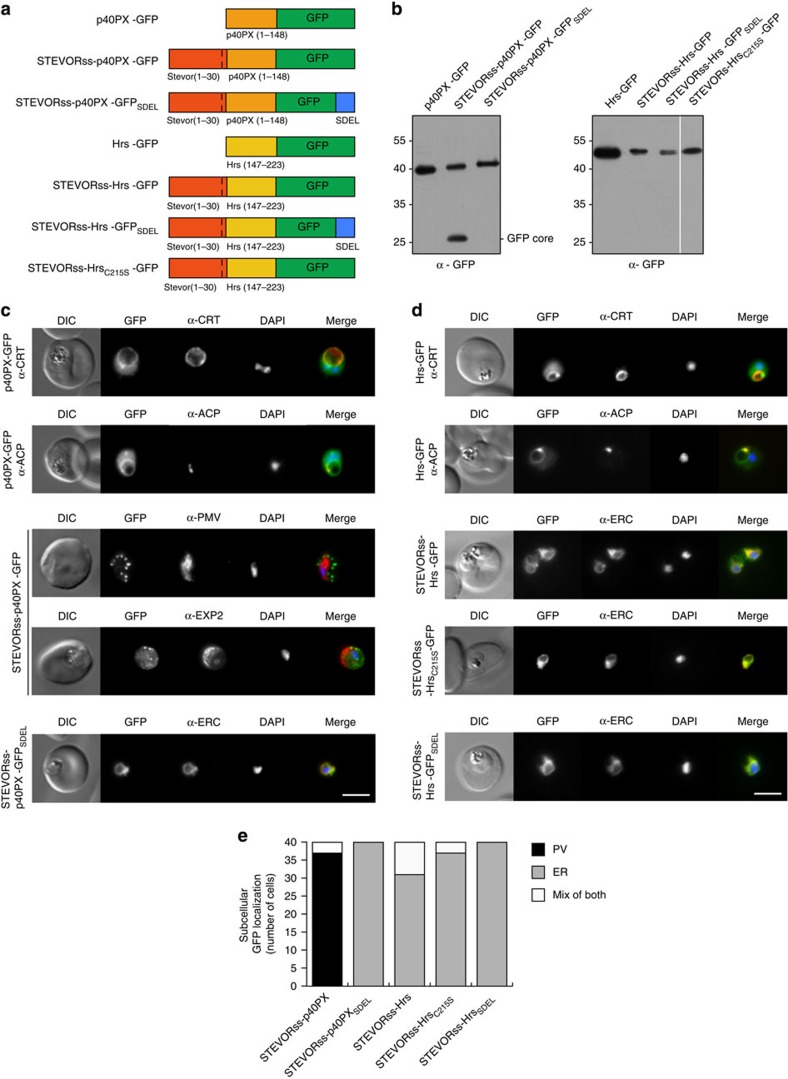
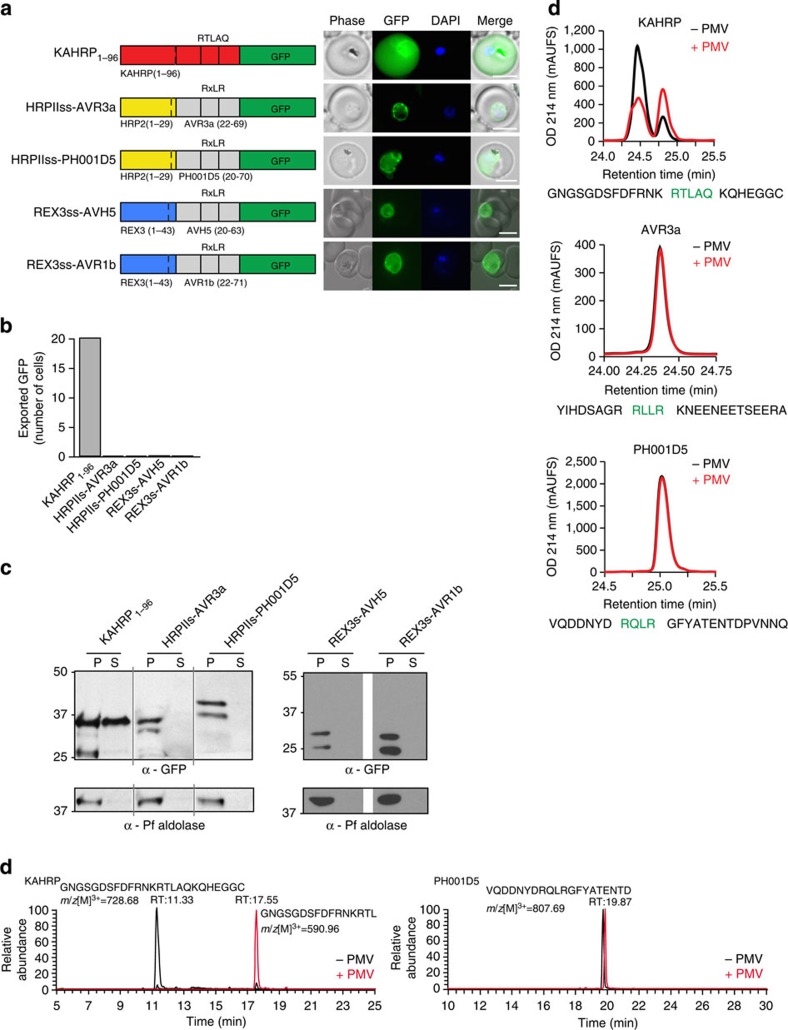
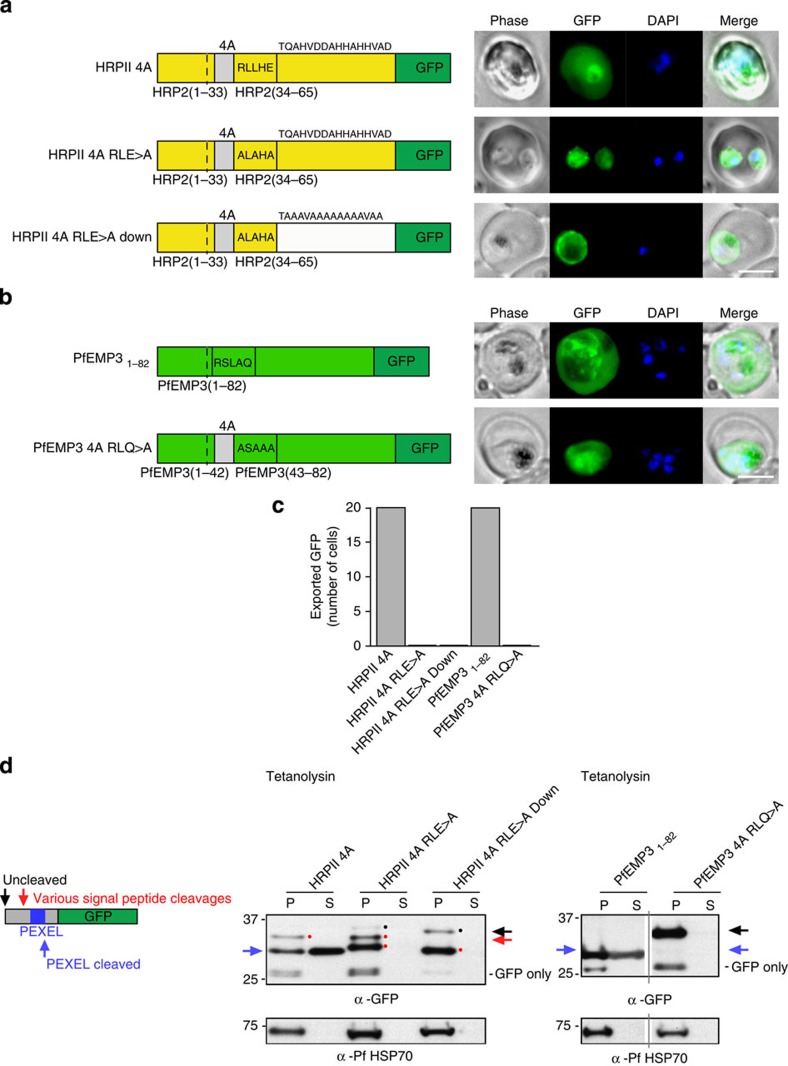
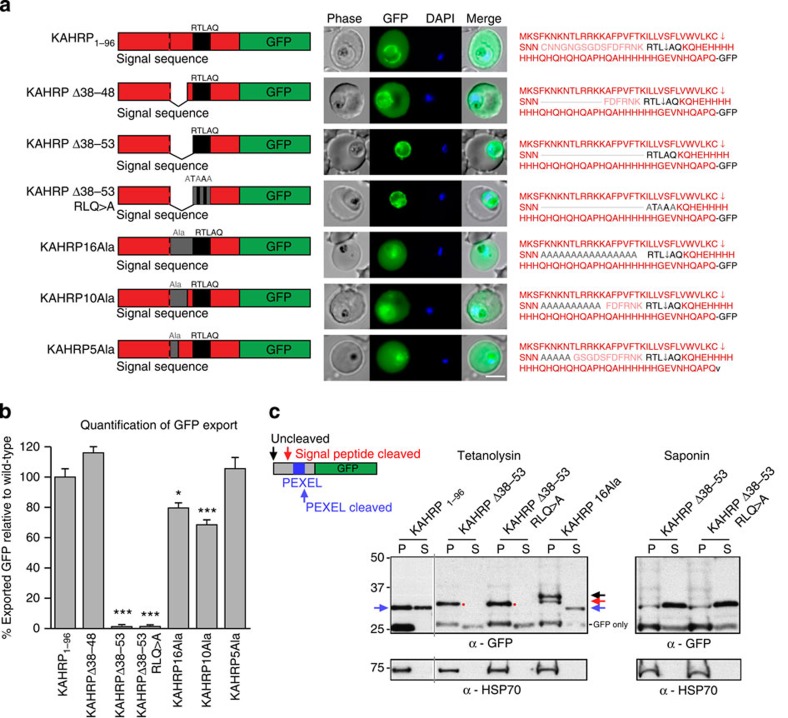
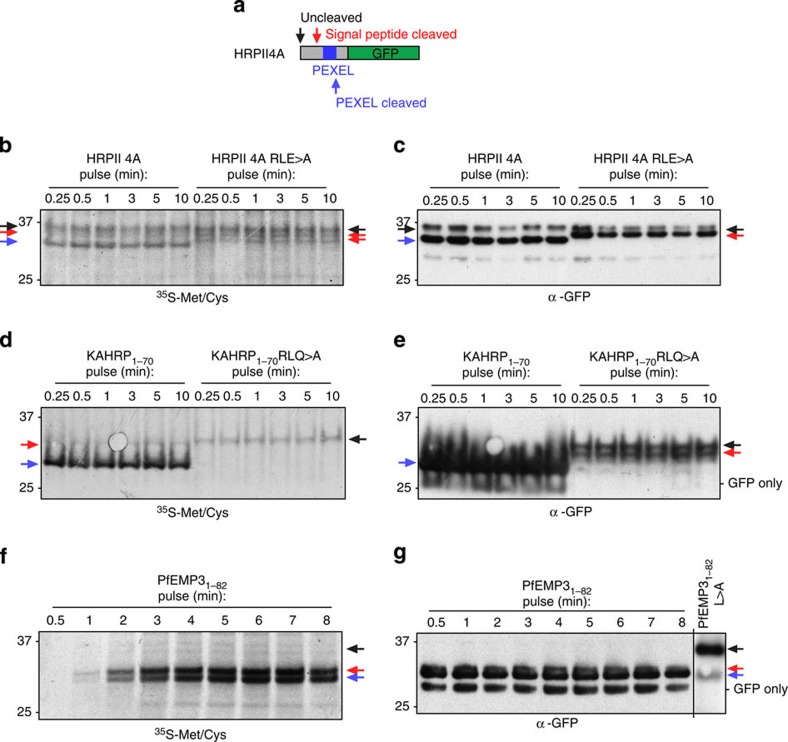
Plasmodium falciparum exports proteins into erythrocytes using the Plasmodium export element (PEXEL) motif, which is cleaved in the endoplasmic reticulum (ER) by plasmepsin V (PMV). A recent study reported that phosphatidylinositol-3-phosphate (PI(3)P) concentrated in the ER binds to PEXEL motifs and is required for export independent of PMV, and that PEXEL motifs are functionally interchangeable with RxLR motifs of oomycete effectors. Here we show that the PEXEL does not bind PI(3)P, and that this lipid is not concentrated in the ER. We find that RxLR motifs cannot mediate export in P. falciparum. Parasites expressing a mutated version of KAHRP, with the PEXEL motif repositioned near the signal sequence, prevented PMV cleavage. This mutant possessed the putative PI(3)P-binding residues but is not exported. Reinstatement of PEXEL to its original location restores processing by PMV and export. These results challenge the PI(3)P hypothesis and provide evidence that PEXEL position is conserved for co-translational processing and export.
Figures
References
-
- Boddey J. A. & Cowman A. F. Plasmodium nesting: remaking the erythrocyte from the inside out. Annu. Rev. Microbiol. 67, 243–269 (2013). - PubMed
-
- Nash G. B., O'Brien E., Gordon-Smith E. C. & Dormandy J. A. Abnormalities in the mechanical properties of red blood cells caused by Plasmodium falciparum. Blood 74, 855–861 (1989). - PubMed
-
- Leech J. H., Aley S. B., Miller L. H. & Howard R. J. Plasmodium falciparum malaria: cytoadherence of infected erythrocytes to endothelial cells and associated changes in the erythrocyte membrane. Prog. Clin. Biol. Res. 155, 63–77 (1984). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
