

The evolutionary turnover of recombination hot spots contributes to speciation in mice
- PMID: 26833728
- PMCID: PMC4743057
- DOI: 10.1101/gad.270009.115
The evolutionary turnover of recombination hot spots contributes to speciation in mice
Erratum in
-
Erratum: The evolutionary turnover of recombination hot spots contributes to speciation in mice.Genes Dev. 2016 Apr 1;30(7):871. doi: 10.1101/gad.280677.116. Genes Dev. 2016. PMID: 27036969 Free PMC article.
-
Corrigendum: The evolutionary turnover of recombination hot spots contributes to speciation in mice.Genes Dev. 2024 Jun 1;38(11-12):583. doi: 10.1101/gad.352049.124. Genes Dev. 2024. PMID: 39029951 Free PMC article. No abstract available.
Abstract
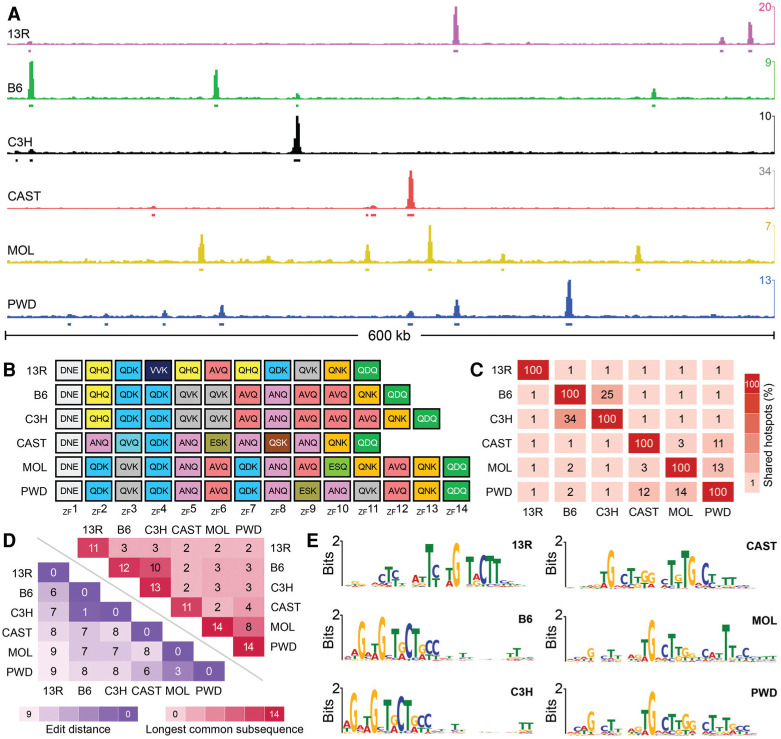
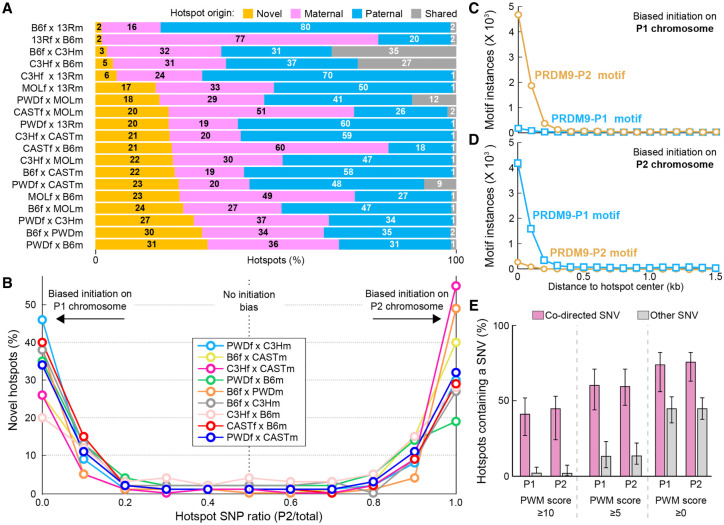
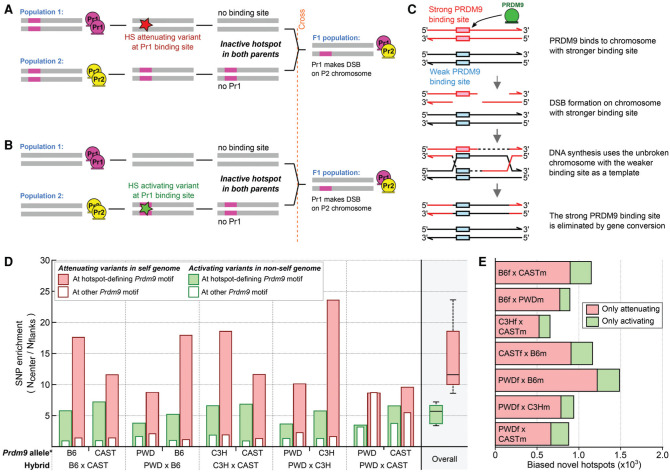
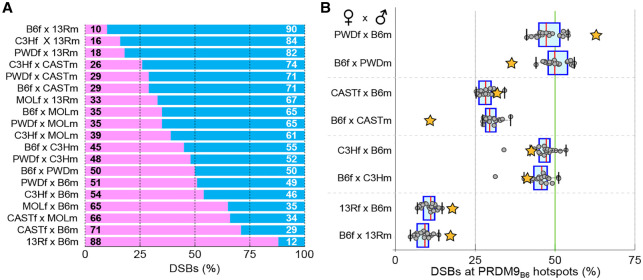
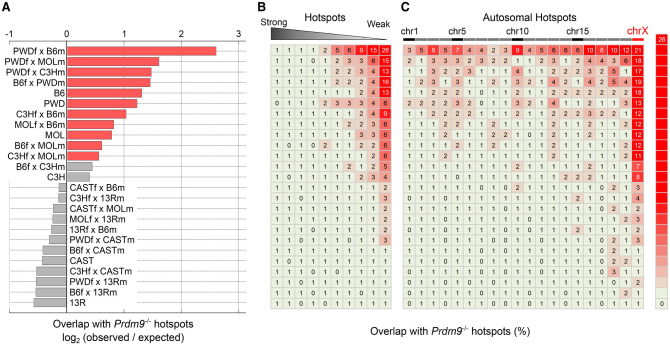
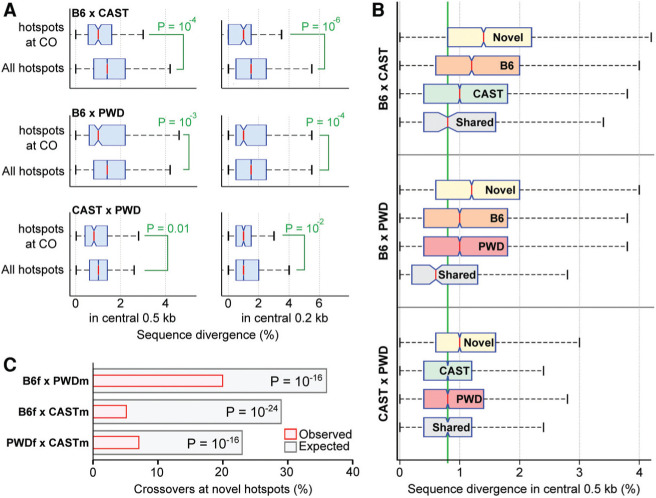
Meiotic recombination is required for the segregation of homologous chromosomes and is essential for fertility. In most mammals, the DNA double-strand breaks (DSBs) that initiate meiotic recombination are directed to a subset of genomic loci (hot spots) by sequence-specific binding of the PRDM9 protein. Rapid evolution of the DNA-binding specificity of PRDM9 and gradual erosion of PRDM9-binding sites by gene conversion will alter the recombination landscape over time. To better understand the evolutionary turnover of recombination hot spots and its consequences, we mapped DSB hot spots in four major subspecies of Mus musculus with different Prdm9 alleles and in their F1 hybrids. We found that hot spot erosion governs the preferential usage of some Prdm9 alleles over others in hybrid mice and increases sequence diversity specifically at hot spots that become active in the hybrids. As crossovers are disfavored at such hot spots, we propose that sequence divergence generated by hot spot turnover may create an impediment for recombination in hybrids, potentially leading to reduced fertility and, eventually, speciation.
Keywords: DSB hot spots; Prdm9; homologous recombination; hybrid sterility; meiosis; recombination hot spots; speciation.
Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Baudat F, Imai Y, de Massy B. 2013. Meiotic recombination in mammals: localization and regulation. Nat Rev Genet 14: 794–806. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials