

Epigenetics and Human Disease
- PMID: 26834142
- PMCID: PMC4743078
- DOI: 10.1101/cshperspect.a019497
Epigenetics and Human Disease
Abstract
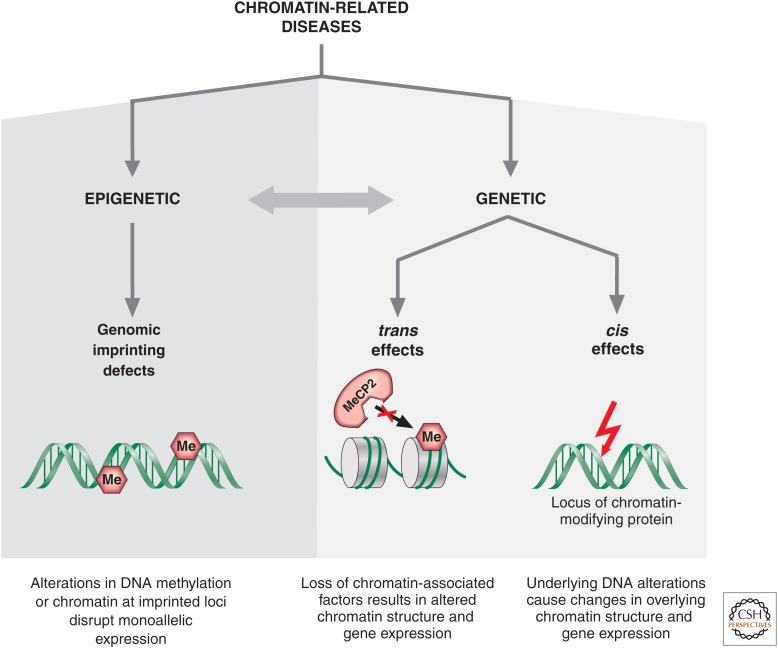
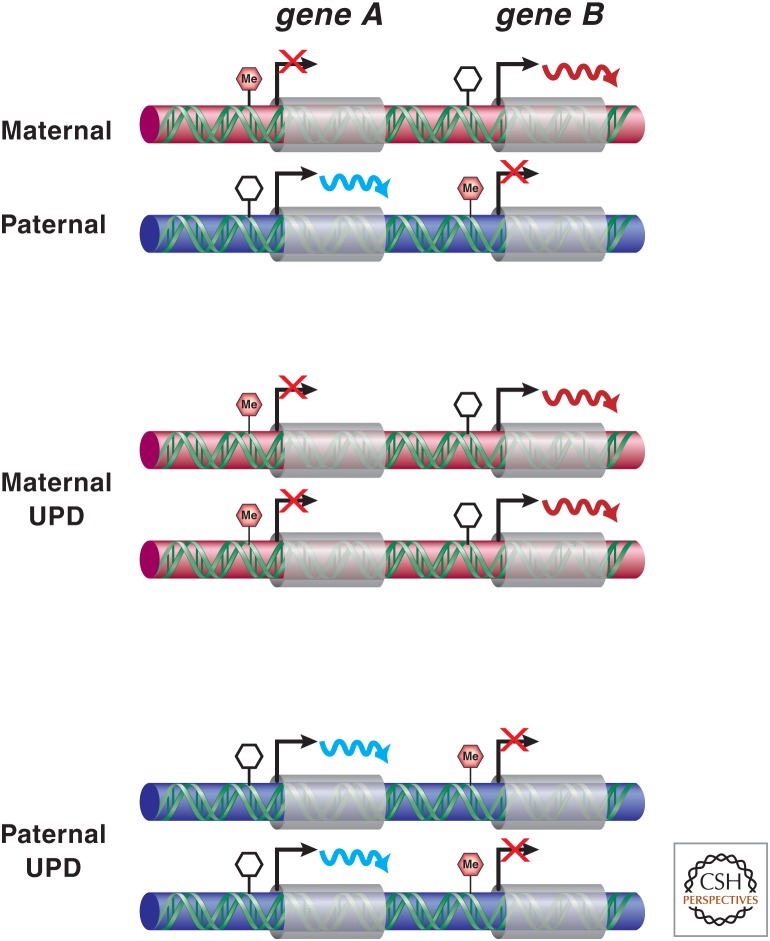
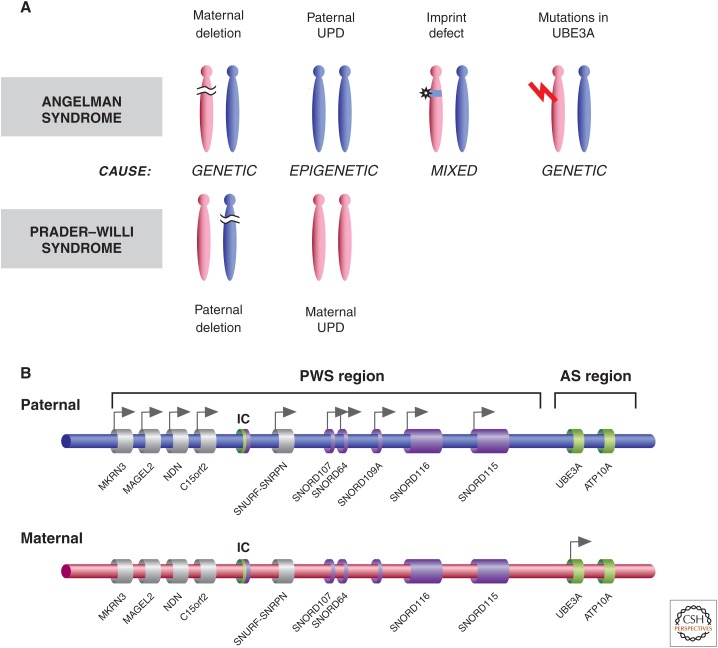
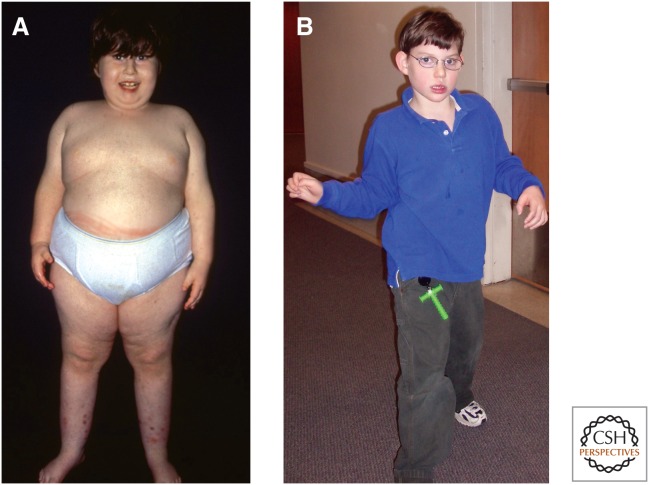
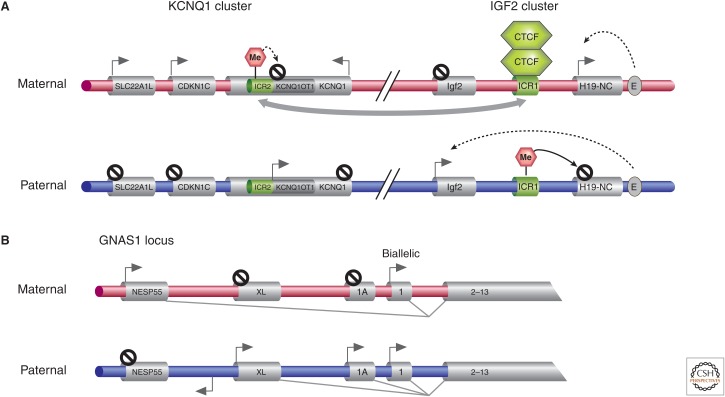
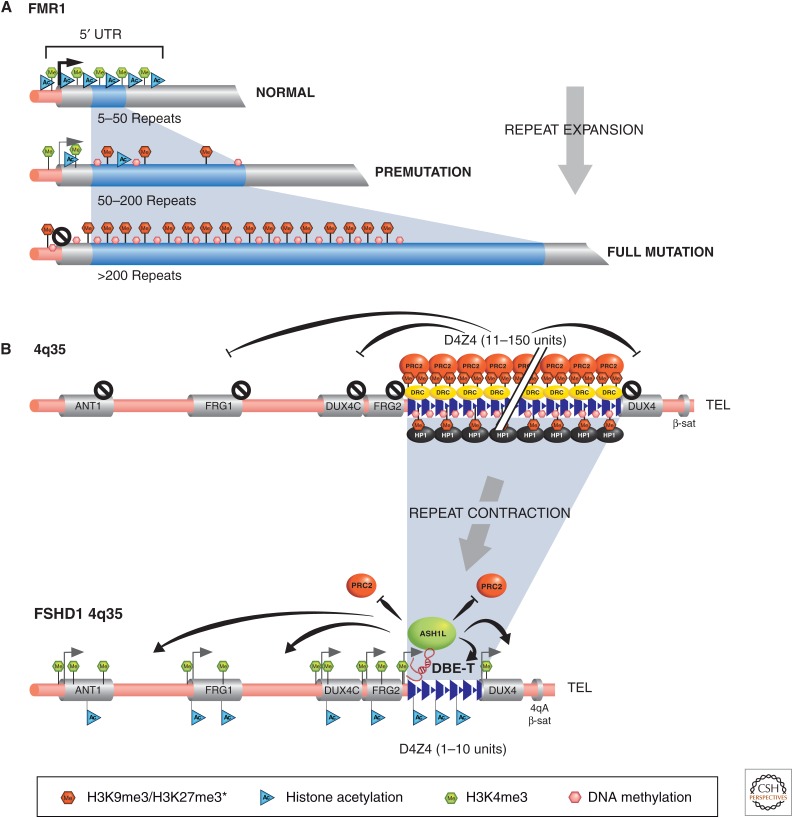
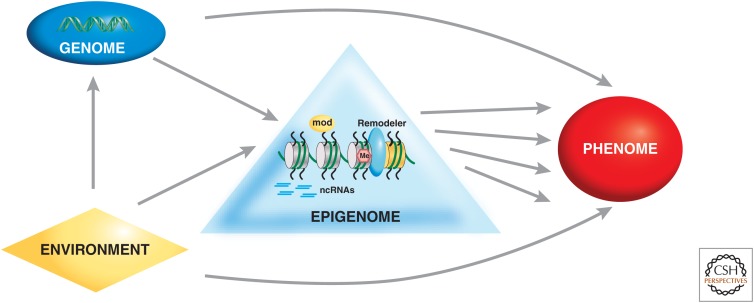
Genetic causes for human disorders are being discovered at an unprecedented pace. A growing subclass of disease-causing mutations involves changes in the epigenome or in the abundance and activity of proteins that regulate chromatin structure. This article focuses on research that has uncovered human diseases that stem from such epigenetic deregulation. Disease may be caused by direct changes in epigenetic marks, such as DNA methylation, commonly found to affect imprinted gene regulation. Also described are disease-causing genetic mutations in epigenetic modifiers that either affect chromatin in trans or have a cis effect in altering chromatin configuration.
Copyright © 2016 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures

Similar articles
-
Epigenetics and human disease.Annu Rev Genomics Hum Genet. 2004;5:479-510. doi: 10.1146/annurev.genom.5.061903.180014. Annu Rev Genomics Hum Genet. 2004. PMID: 15485357 Review.
-
[Epigenetics of schizophrenia: a review].Encephale. 2014 Oct;40(5):380-6. doi: 10.1016/j.encep.2014.06.005. Epub 2014 Aug 12. Encephale. 2014. PMID: 25127897 Review. French.
-
Zebrafish Discoveries in Cancer Epigenetics.Adv Exp Med Biol. 2016;916:169-97. doi: 10.1007/978-3-319-30654-4_8. Adv Exp Med Biol. 2016. PMID: 27165354 Free PMC article. Review.
-
Cell Type-Specific Chromatin Signatures Underline Regulatory DNA Elements in Human Induced Pluripotent Stem Cells and Somatic Cells.Circ Res. 2017 Nov 10;121(11):1237-1250. doi: 10.1161/CIRCRESAHA.117.311367. Epub 2017 Oct 13. Circ Res. 2017. PMID: 29030344 Free PMC article.
-
Mammalian epigenetic mechanisms.IUBMB Life. 2014 Apr;66(4):240-56. doi: 10.1002/iub.1264. Epub 2014 Apr 5. IUBMB Life. 2014. PMID: 24706538 Review.
Cited by
-
mir152 hypomethylation as a mechanism for non-syndromic cleft lip and palate.Epigenetics. 2022 Dec;17(13):2278-2295. doi: 10.1080/15592294.2022.2115606. Epub 2022 Sep 1. Epigenetics. 2022. PMID: 36047706 Free PMC article.
-
Epigenetic Determinants of Cancer.Cold Spring Harb Perspect Biol. 2016 Sep 1;8(9):a019505. doi: 10.1101/cshperspect.a019505. Cold Spring Harb Perspect Biol. 2016. PMID: 27194046 Free PMC article. Review.
-
Deep Learning for Human Disease Detection, Subtype Classification, and Treatment Response Prediction Using Epigenomic Data.Biomedicines. 2021 Nov 20;9(11):1733. doi: 10.3390/biomedicines9111733. Biomedicines. 2021. PMID: 34829962 Free PMC article. Review.
-
Direct measurement of the mechanical properties of a chromatin analog and the epigenetic effects of para-sulphonato-calix[4]arene.Sci Rep. 2019 Apr 9;9(1):5816. doi: 10.1038/s41598-019-42267-x. Sci Rep. 2019. PMID: 30967623 Free PMC article.
-
DNA methylome signatures as epigenetic biomarkers of hexanal associated with lung toxicity.PeerJ. 2021 Feb 4;9:e10779. doi: 10.7717/peerj.10779. eCollection 2021. PeerJ. 2021. PMID: 33604181 Free PMC article.
References
-
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. 2004. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: A model for the cognitive deficit in Rubinstein–Taybi syndrome and its amelioration. Neuron 42: 947–959. - PubMed
-
- Allis CD, Jenuwein T, Reinberg D, 2014. Overview and concepts. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a018739. - DOI
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources