

Evasion of the Immune Response by Trypanosoma cruzi during Acute Infection
- PMID: 26834737
- PMCID: PMC4716143
- DOI: 10.3389/fimmu.2015.00659
Evasion of the Immune Response by Trypanosoma cruzi during Acute Infection
Abstract
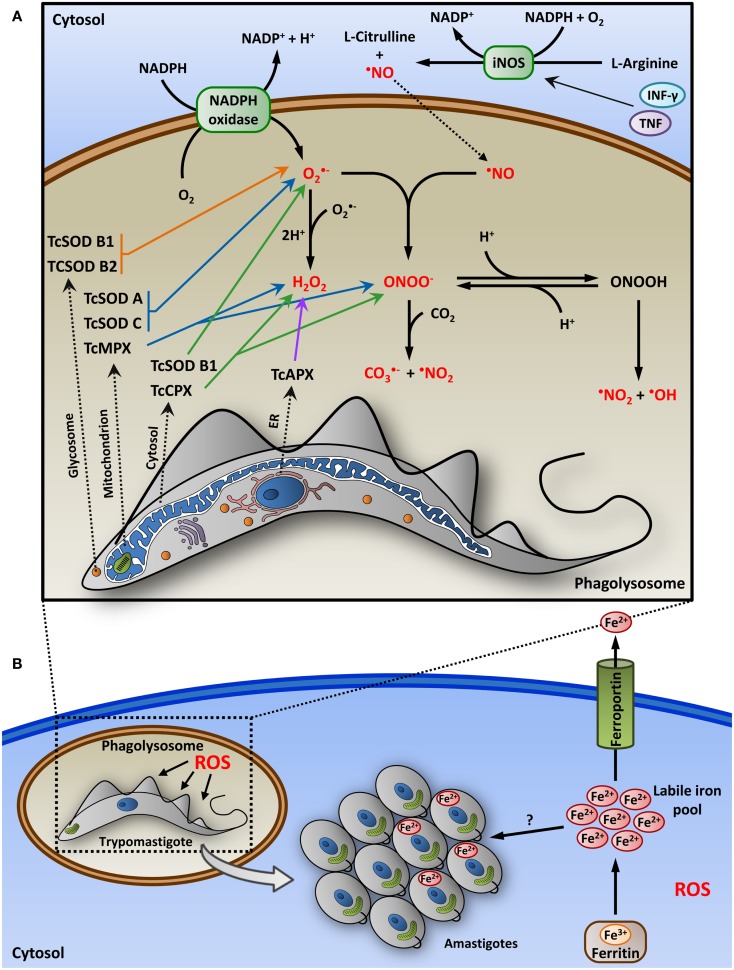
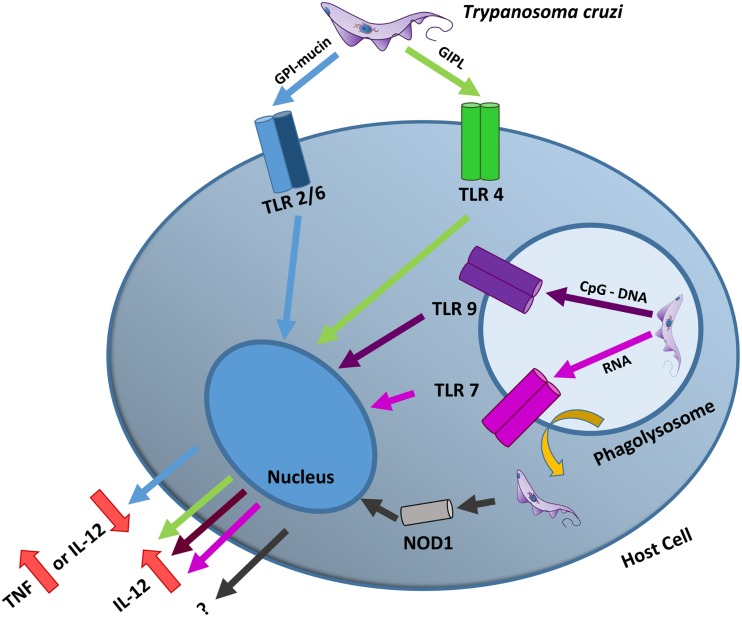
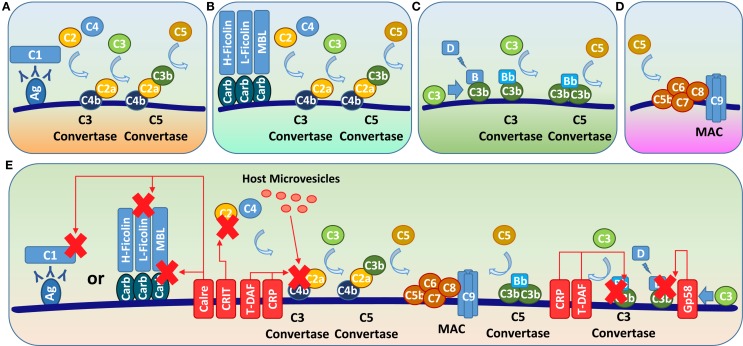
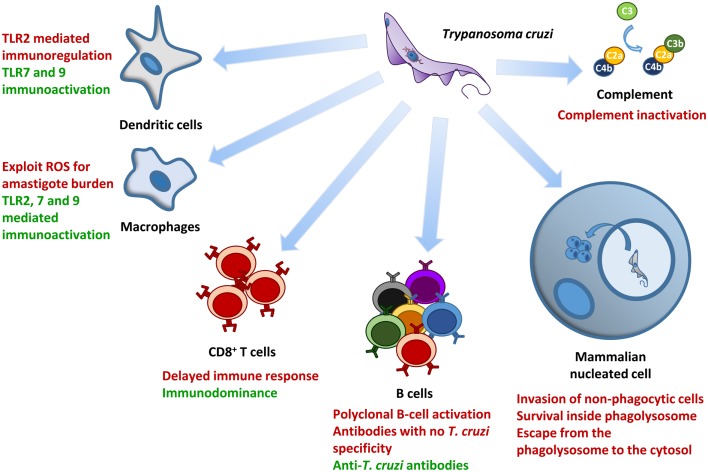
Trypanosoma cruzi is the etiologic agent of Chagas disease, a neglected tropical disease that affects millions of people mainly in Latin America. To establish a life-long infection, T. cruzi must subvert the vertebrate host's immune system, using strategies that can be traced to the parasite's life cycle. Once inside the vertebrate host, metacyclic trypomastigotes rapidly invade a wide variety of nucleated host cells in a membrane-bound compartment known as the parasitophorous vacuole, which fuses to lysosomes, originating the phagolysosome. In this compartment, the parasite relies on a complex network of antioxidant enzymes to shield itself from lysosomal oxygen and nitrogen reactive species. Lysosomal acidification of the parasitophorous vacuole is an important factor that allows trypomastigote escape from the extremely oxidative environment of the phagolysosome to the cytoplasm, where it differentiates into amastigote forms. In the cytosol of infected macrophages, oxidative stress instead of being detrimental to the parasite, favors amastigote burden, which then differentiates into bloodstream trypomastigotes. Trypomastigotes released in the bloodstream upon the rupture of the host cell membrane express surface molecules, such as calreticulin and GP160 proteins, which disrupt initial and key components of the complement pathway, while others such as glycosylphosphatidylinositol-mucins stimulate immunoregulatory receptors, delaying the progression of a protective immune response. After an immunologically silent entry at the early phase of infection, T. cruzi elicits polyclonal B cell activation, hypergammaglobulinemia, and unspecific anti-T. cruzi antibodies, which are inefficient in controlling the infection. Additionally, the coexpression of several related, but not identical, epitopes derived from trypomastigote surface proteins delays the generation of T. cruzi-specific neutralizing antibodies. Later in the infection, the establishment of an anti-T. cruzi CD8(+) immune response focused on the parasite's immunodominant epitopes controls parasitemia and tissue infection, but fails to completely eliminate the parasite. This outcome is not detrimental to the parasite, as it reduces host mortality and maintains the parasite infectivity toward the insect vectors.
Keywords: Chagas disease; T. cruzi acute infection; T. cruzi immune evasion; immune response; immunomodulation.
Figures
References
-
- Hotez PJ, Bottazzi ME, Franco-Paredes C, Ault SK, Periago MR. The neglected tropical diseases of Latin America and the Caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS Negl Trop Dis (2008) 2(9):e300. 10.1371/journal.pntd.0000300 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials