

Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies
- PMID: 26834781
- PMCID: PMC4719219
- DOI: 10.3389/fgene.2015.00359
Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies
Abstract
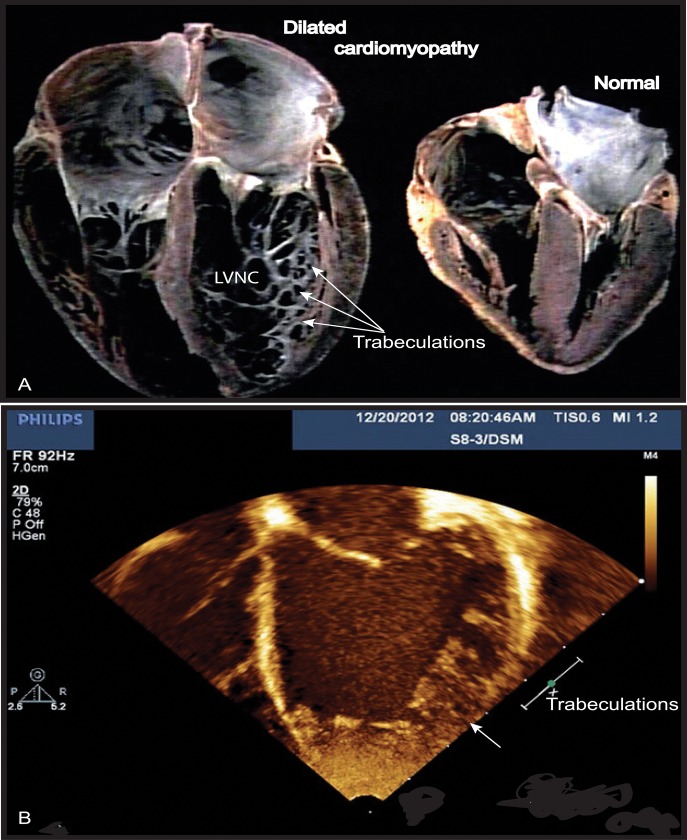
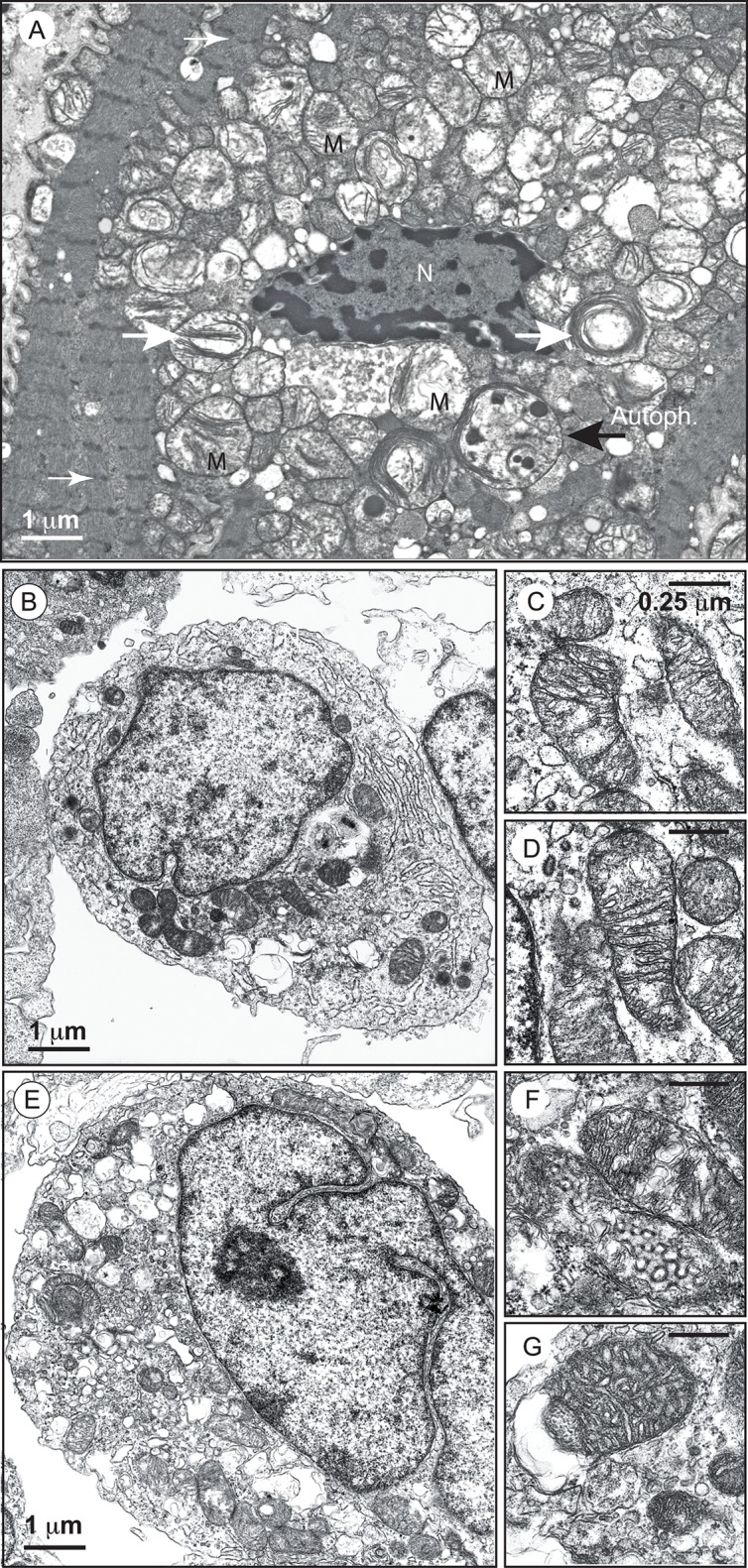
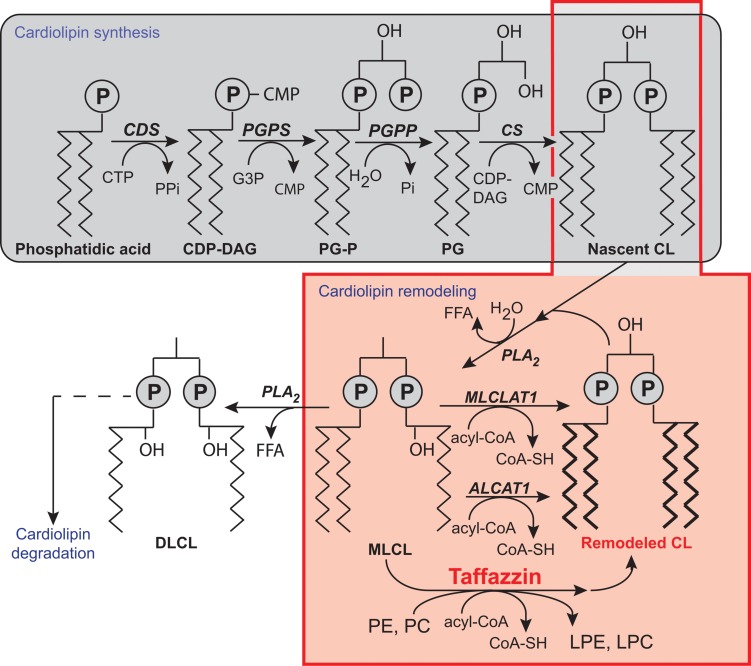
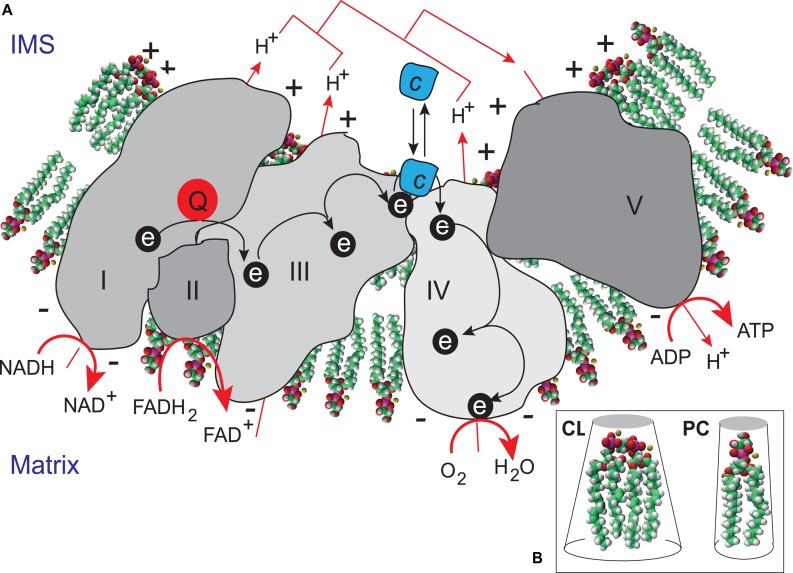
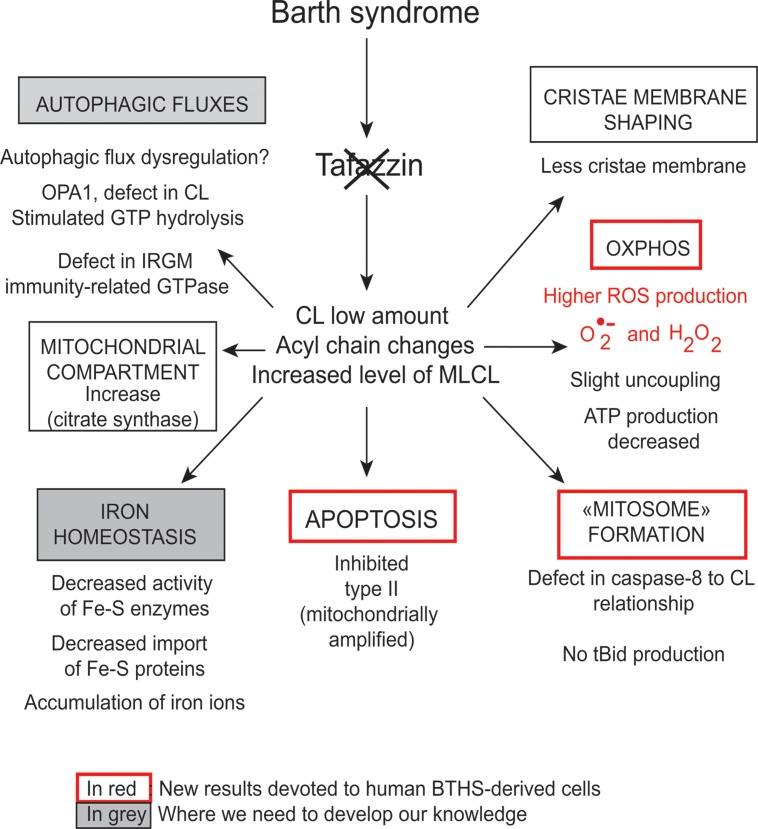
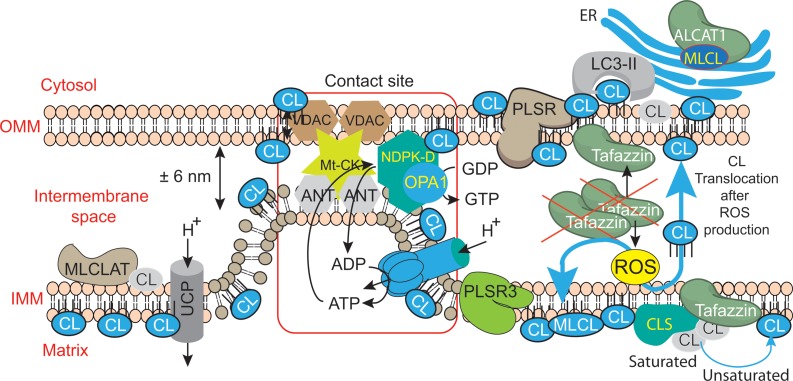
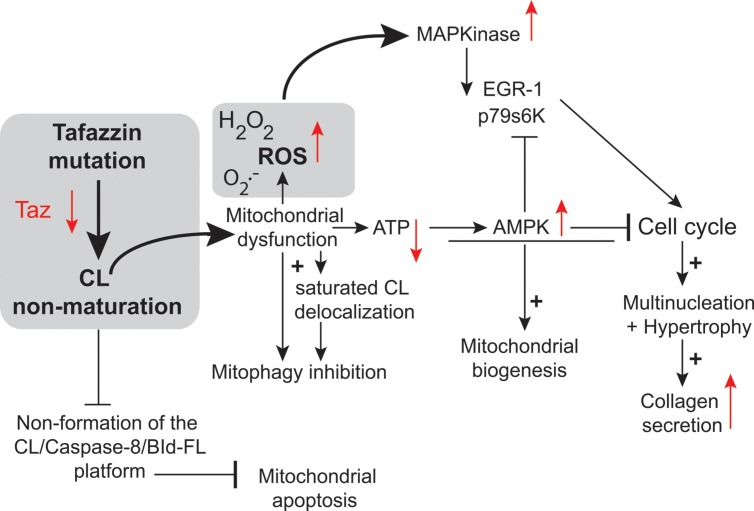
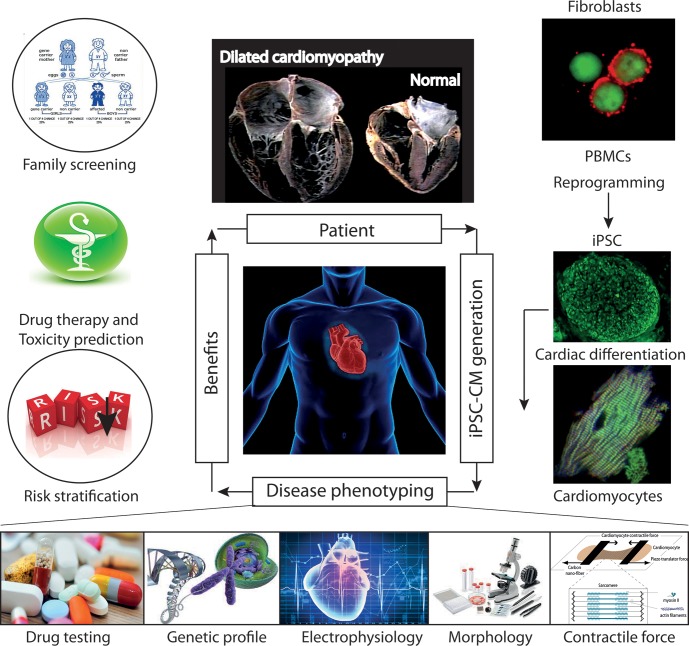
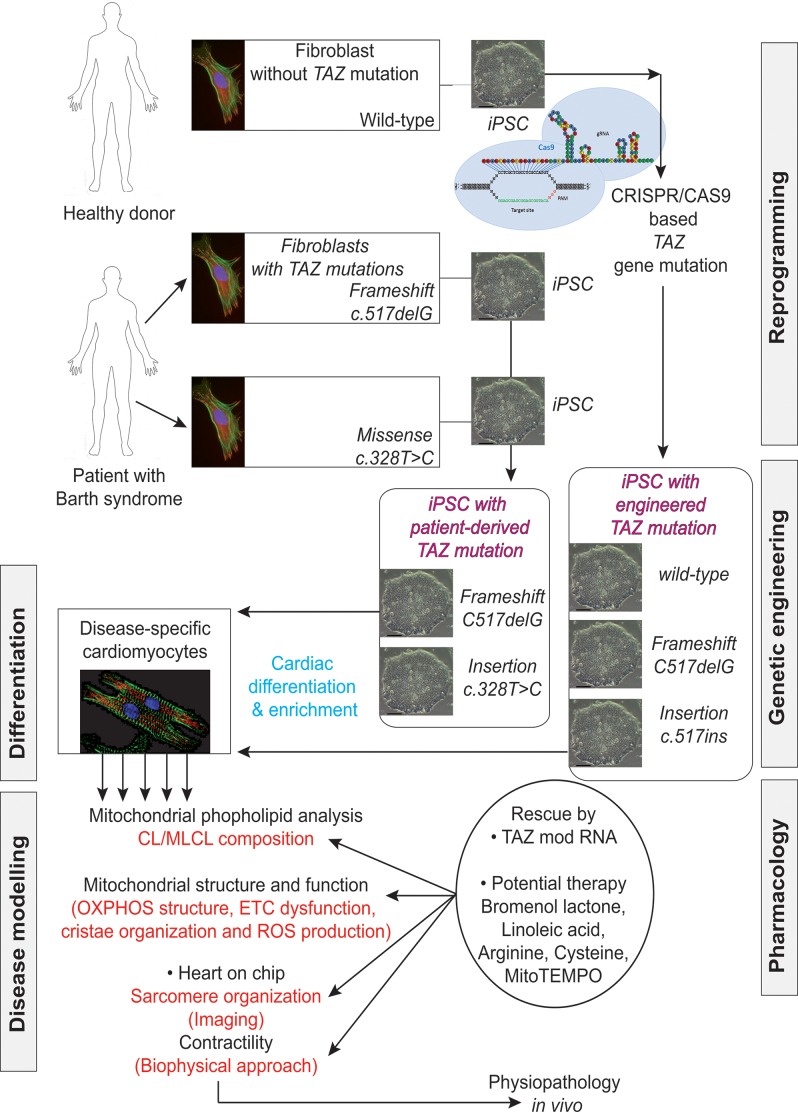
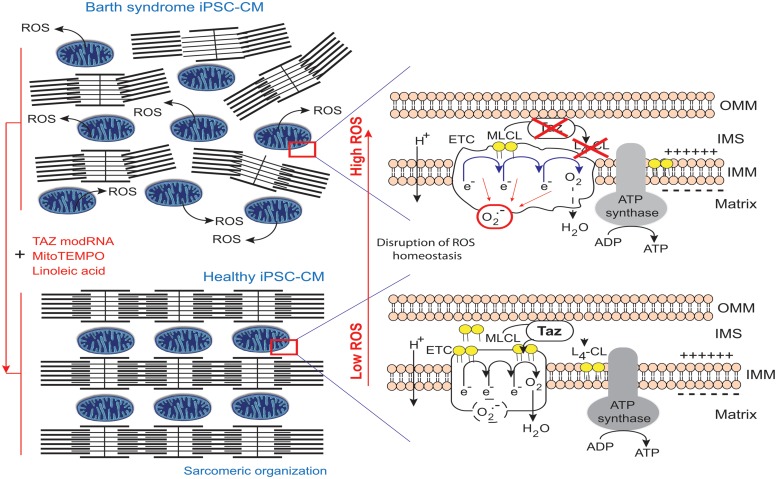
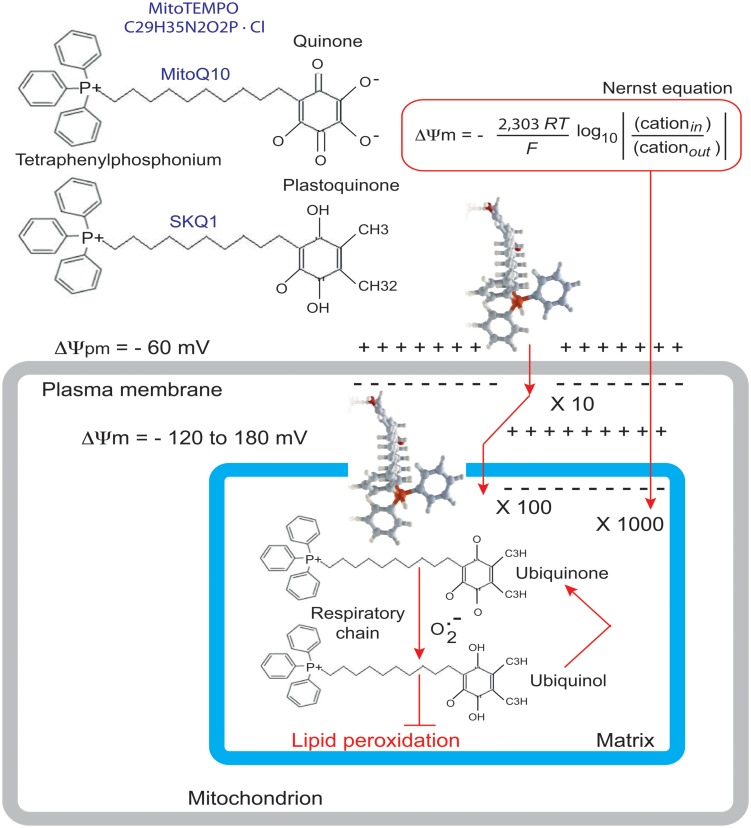
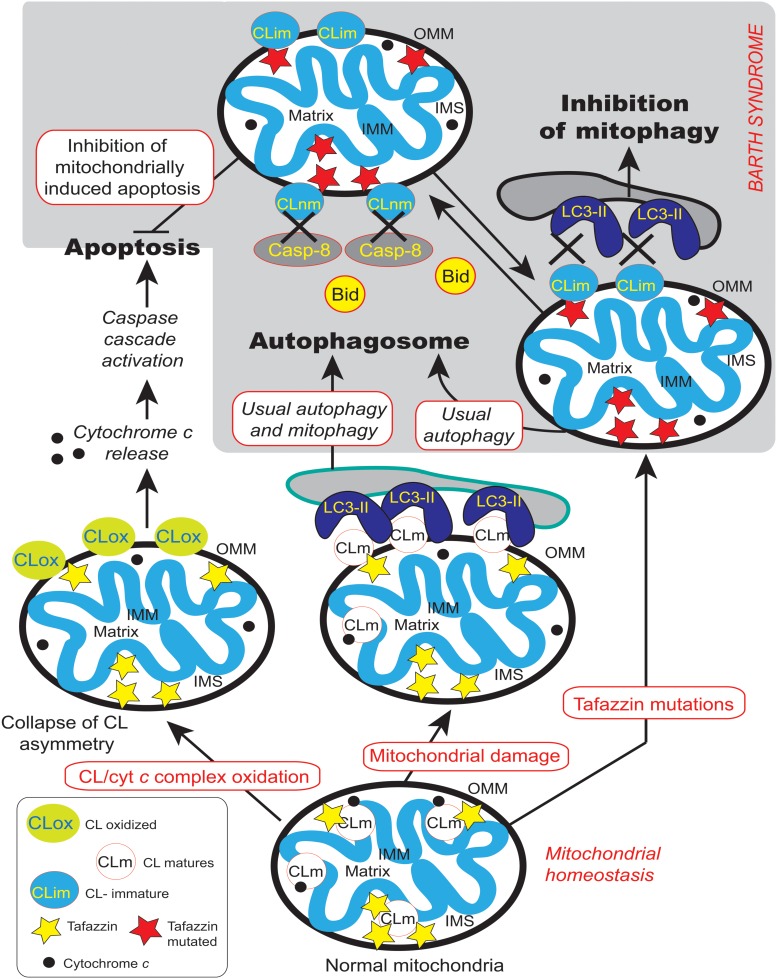
Mutations in the gene encoding the enzyme tafazzin, TAZ, cause Barth syndrome (BTHS). Individuals with this X-linked multisystem disorder present cardiomyopathy (CM) (often dilated), skeletal muscle weakness, neutropenia, growth retardation, and 3-methylglutaconic aciduria. Biopsies of the heart, liver and skeletal muscle of patients have revealed mitochondrial malformations and dysfunctions. It is the purpose of this review to summarize recent results of studies on various animal or cell models of Barth syndrome, which have characterized biochemically the strong cellular defects associated with TAZ mutations. Tafazzin is a mitochondrial phospholipidlysophospholipid transacylase that shuttles acyl groups between phospholipids and regulates the remodeling of cardiolipin (CL), a unique inner mitochondrial membrane phospholipid dimer consisting of two phosphatidyl residues linked by a glycerol bridge. After their biosynthesis, the acyl chains of CLs may be modified in remodeling processes involving up to three different enzymes. Their characteristic acyl chain composition depends on the function of tafazzin, although the enzyme itself surprisingly lacks acyl specificity. CLs are crucial for correct mitochondrial structure and function. In addition to their function in the basic mitochondrial function of ATP production, CLs play essential roles in cardiac function, apoptosis, autophagy, cell cycle regulation and Fe-S cluster biosynthesis. Recent developments in tafazzin research have provided strong insights into the link between mitochondrial dysfunction and the production of reactive oxygen species (ROS). An important tool has been the generation of BTHS-specific induced pluripotent stem cells (iPSCs) from BTHS patients. In a complementary approach, disease-specific mutations have been introduced into wild-type iPSC lines enabling direct comparison with isogenic controls. iPSC-derived cardiomyocytes were then characterized using biochemical and classical bioenergetic approaches. The cells are tested in a "heart-on-chip" assay to model the pathophysiology in vitro, to characterize the underlying mechanism of BTHS deriving from TAZ mutations, mitochondrial deficiencies and ROS production and leading to tissue defects, and to evaluate potential therapies with the use of mitochondrially targeted antioxidants.
Keywords: barth syndrome; cardiolipin; cellular models; mitochondria; mitochondrially targeted antioxidant; stem cells; tafazzin.
Figures
Similar articles
-
Tafazzin deficiency impairs CoA-dependent oxidative metabolism in cardiac mitochondria.J Biol Chem. 2020 Aug 28;295(35):12485-12497. doi: 10.1074/jbc.RA119.011229. Epub 2020 Jul 14. J Biol Chem. 2020. PMID: 32665401 Free PMC article.
-
Granulopoietic Dysregulation in a Patient-Tailored Mouse Model of Barth Syndrome.Stem Cell Rev Rep. 2025 Aug 5. doi: 10.1007/s12015-025-10945-1. Online ahead of print. Stem Cell Rev Rep. 2025. PMID: 40762880
-
Increased Reactive Oxygen Species-Mediated Ca2+/Calmodulin-Dependent Protein Kinase II Activation Contributes to Calcium Handling Abnormalities and Impaired Contraction in Barth Syndrome.Circulation. 2021 May 11;143(19):1894-1911. doi: 10.1161/CIRCULATIONAHA.120.048698. Epub 2021 Apr 1. Circulation. 2021. PMID: 33793303 Free PMC article.
-
Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy.Lipids. 2017 Feb;52(2):99-108. doi: 10.1007/s11745-016-4229-7. Epub 2017 Jan 9. Lipids. 2017. PMID: 28070695 Free PMC article. Review.
-
Barth syndrome cardiomyopathy: targeting the mitochondria with elamipretide.Heart Fail Rev. 2021 Mar;26(2):237-253. doi: 10.1007/s10741-020-10031-3. Epub 2020 Oct 1. Heart Fail Rev. 2021. PMID: 33001359 Free PMC article. Review.
Cited by
-
Mieap forms membrane-less organelles involved in cardiolipin metabolism.iScience. 2024 Jan 17;27(2):108916. doi: 10.1016/j.isci.2024.108916. eCollection 2024 Feb 16. iScience. 2024. PMID: 38322995 Free PMC article.
-
Mechanical characterization of isolated mitochondria under conditions of oxidative stress.Biomicrofluidics. 2022 Nov 17;16(6):064101. doi: 10.1063/5.0111581. eCollection 2022 Dec. Biomicrofluidics. 2022. PMID: 36406339 Free PMC article.
-
Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes.Metabolites. 2019 Dec 17;9(12):306. doi: 10.3390/metabo9120306. Metabolites. 2019. PMID: 31861102 Free PMC article.
-
Use of Elamipretide in patients assigned treatment in the compassionate use program: Case series in pediatric patients with rare orphan diseases.JIMD Rep. 2022 Sep 21;64(1):65-70. doi: 10.1002/jmd2.12335. eCollection 2023 Jan. JIMD Rep. 2022. PMID: 36636586 Free PMC article.
-
Elamipretide Promotes Mitophagosome Formation and Prevents Its Reduction Induced by Nutrient Excess in INS1 β-cells.J Mol Biol. 2018 Dec 7;430(24):4823-4833. doi: 10.1016/j.jmb.2018.10.020. Epub 2018 Oct 30. J Mol Biol. 2018. PMID: 30389435 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous